75 ml TAE with 0.6 g Agarose and 7.5 ul EtBr.
Loaded the wells with 10 ul 100 bp ladder and 15 ul DNA loading solution (Mix of 15 ul DNA and 3 ul loading dye).
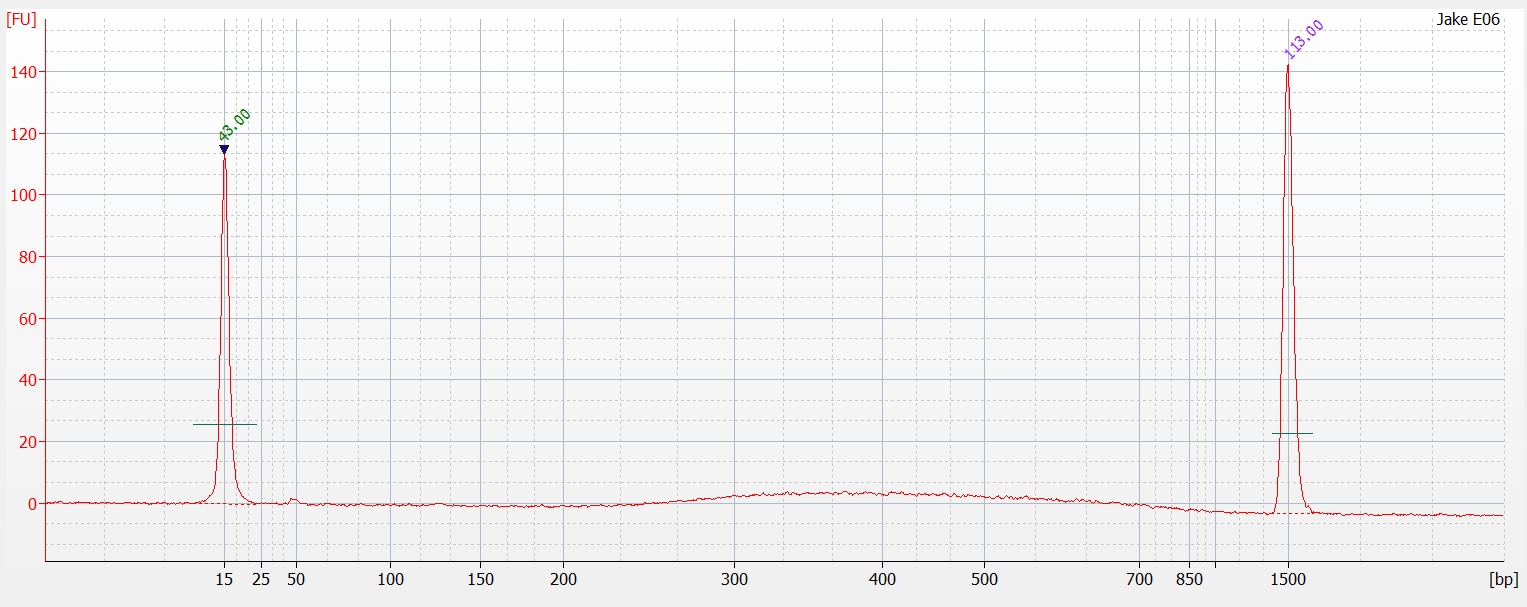
| Well | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Technique | Centrifuged | Spooled | ||||||||||
| Sample | Ladder | Test Oly 1 | Test Oly 2 | Test Oly 3 | Test Oly 4 | Test Oly 1 | Test Oly 2 | Test Oly 3 | Test Oly 4 | Ladder | Empty | Empty |
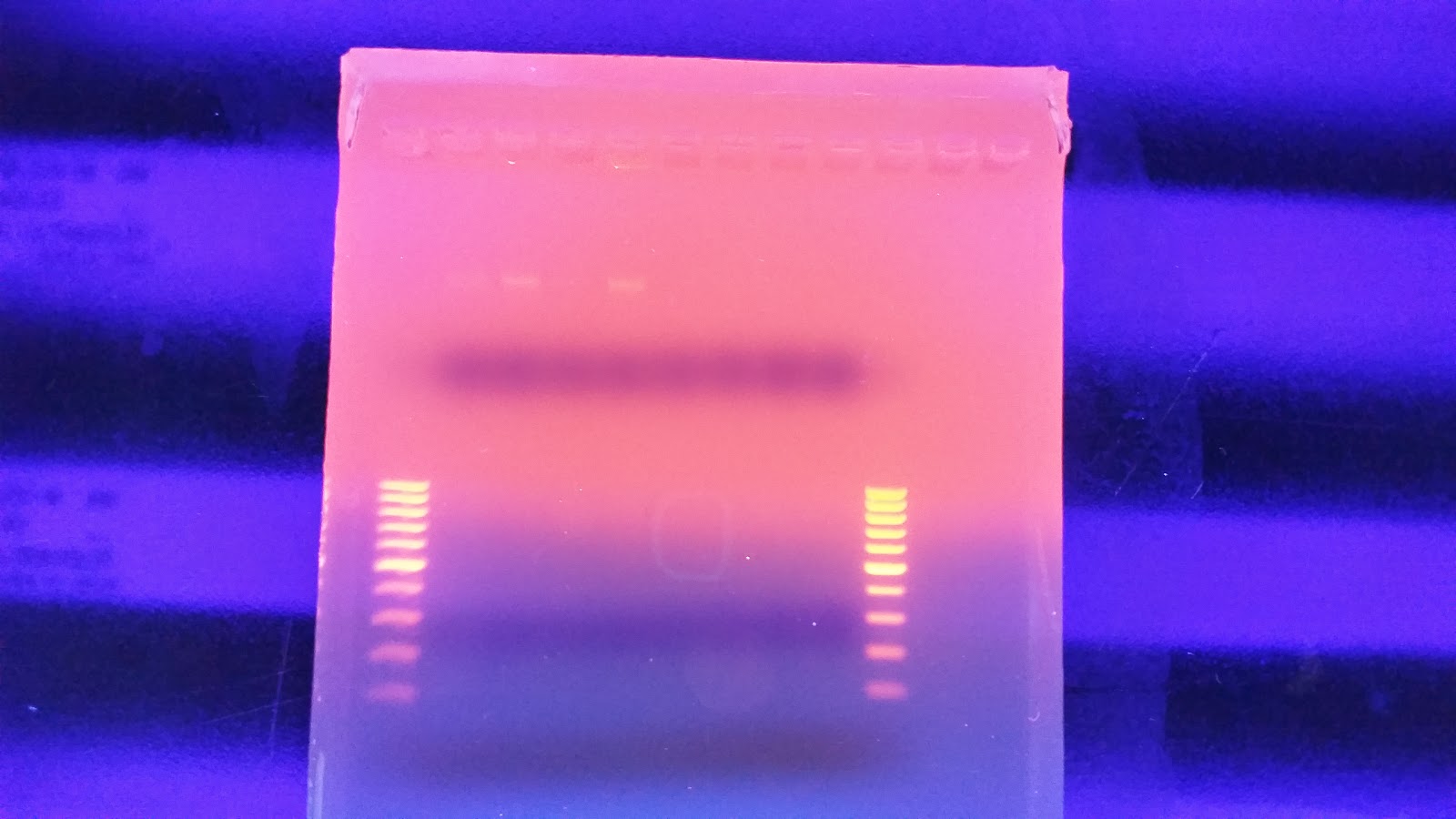 |
| Gel with Fresh Tissue Isolation |
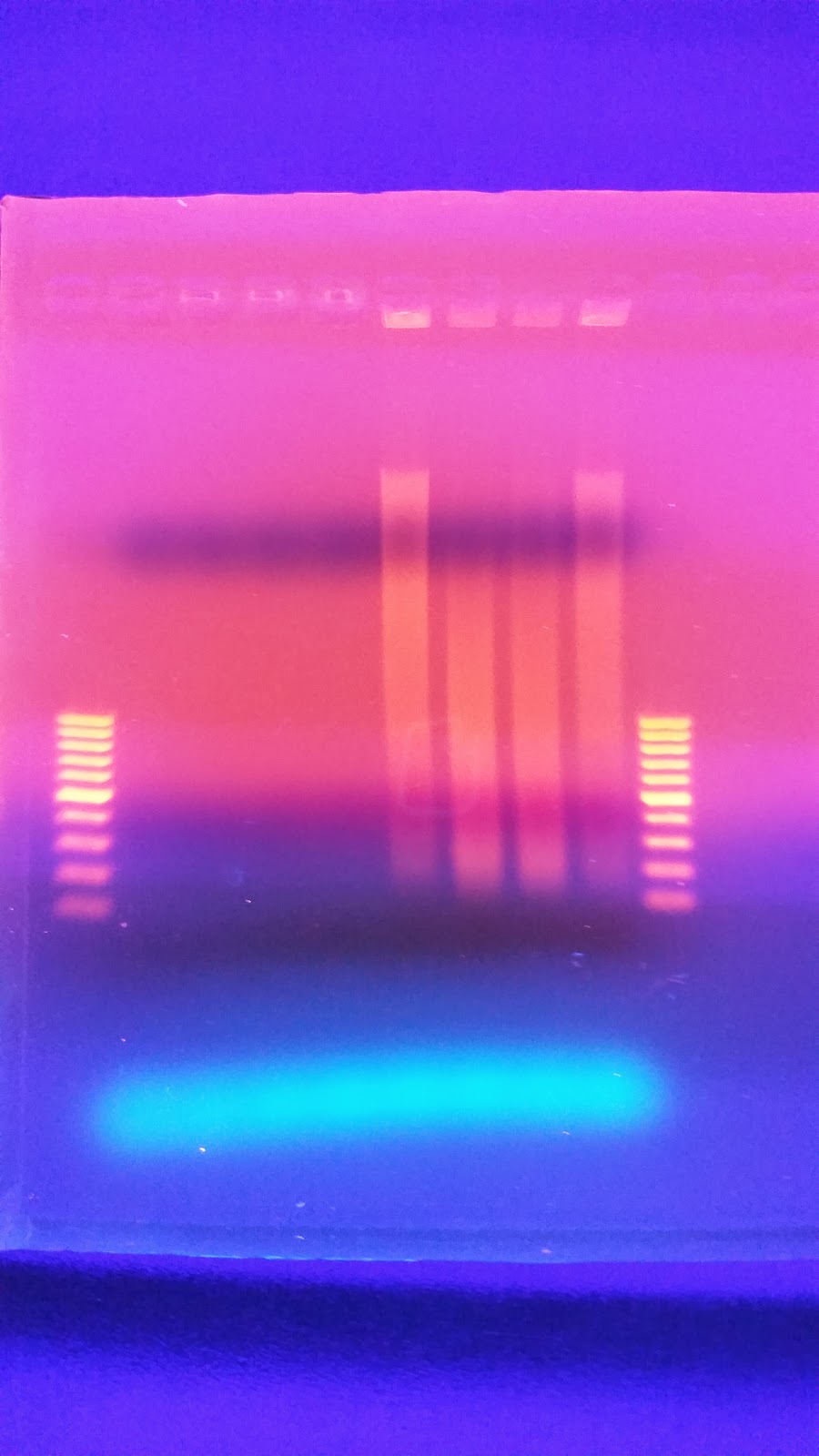
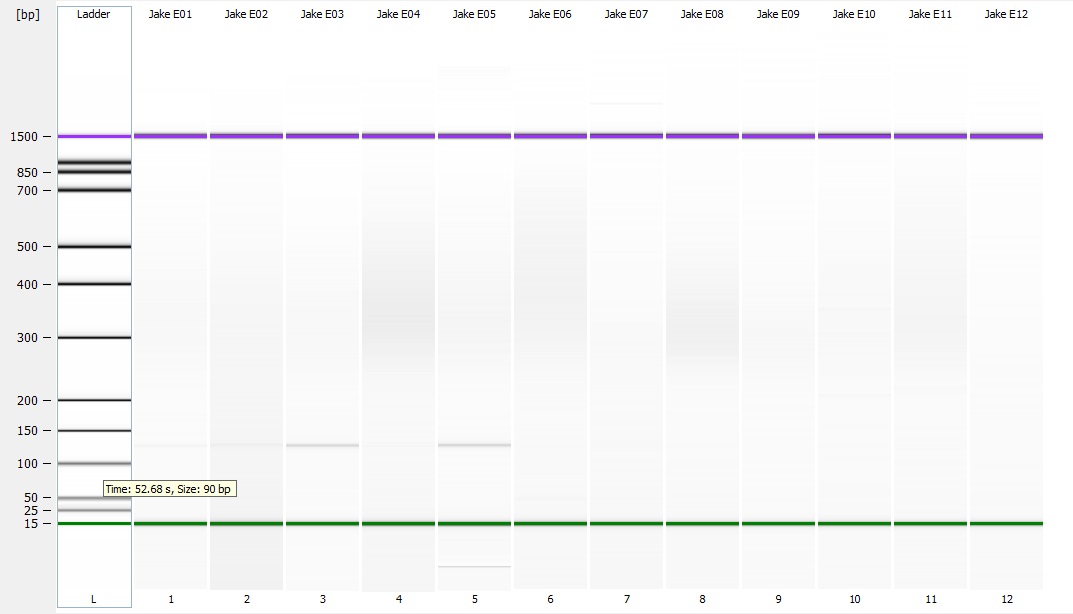
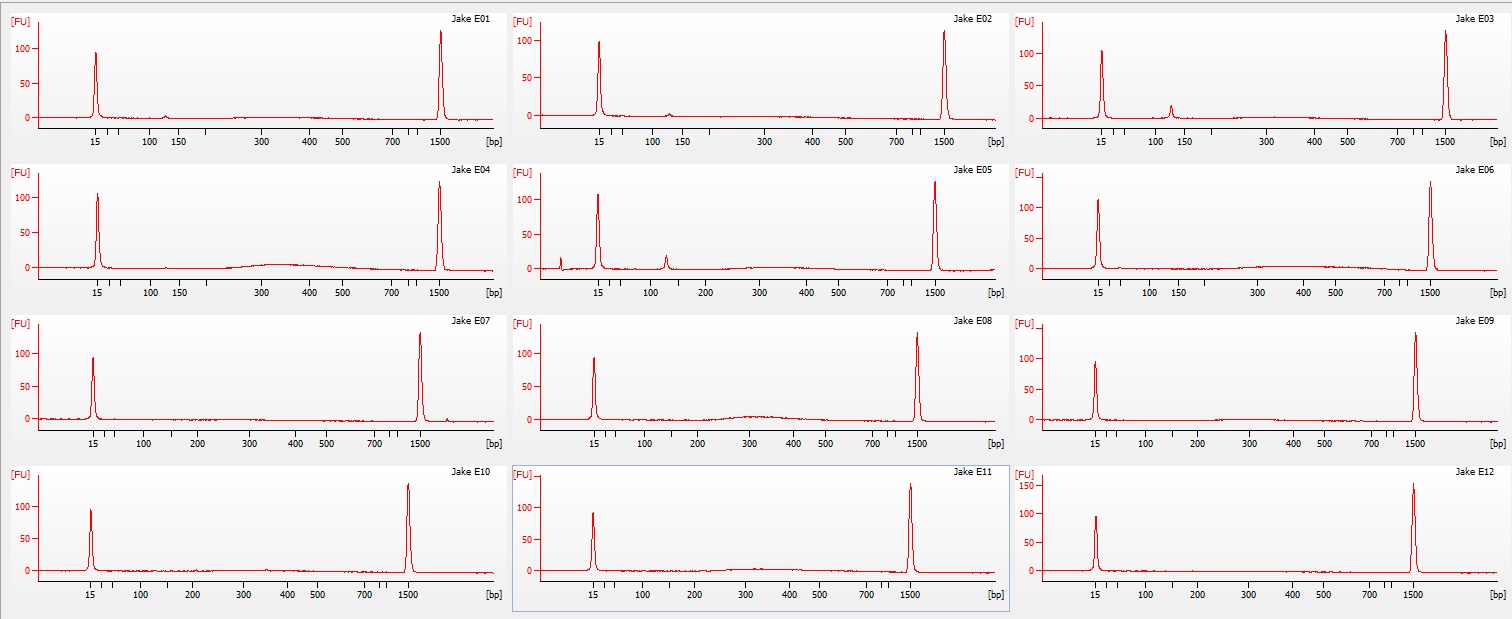
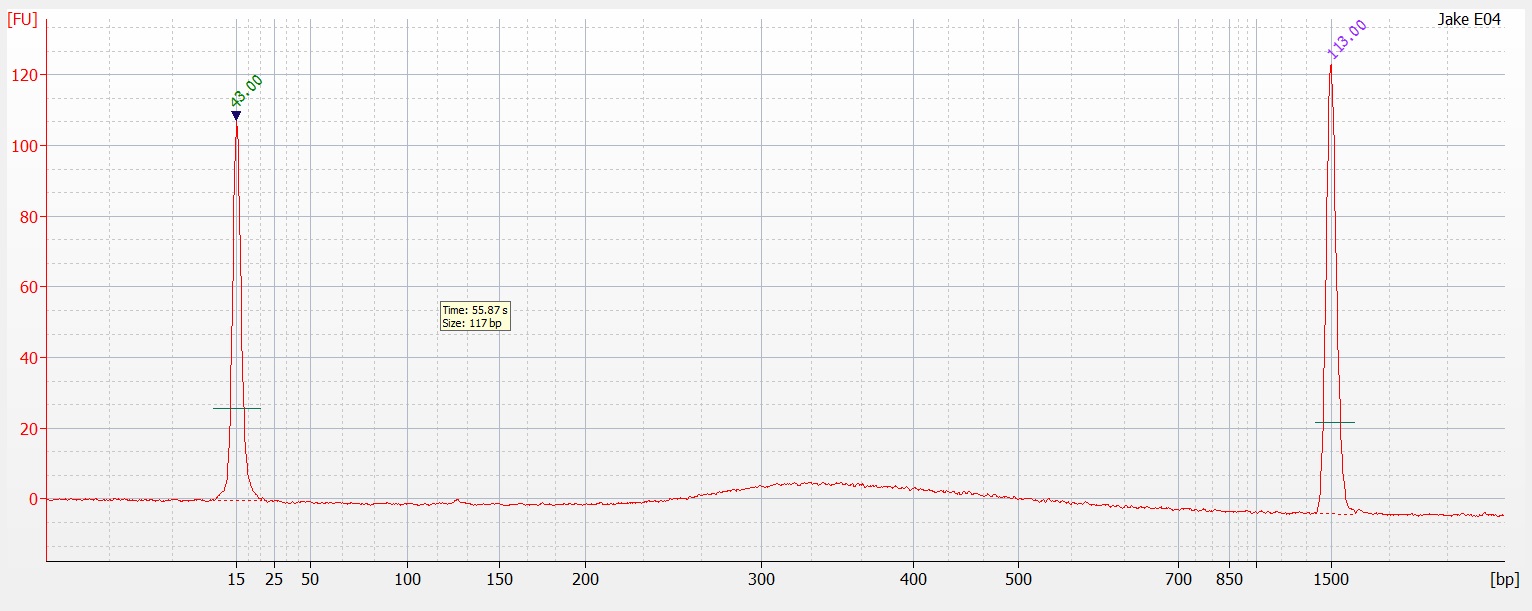
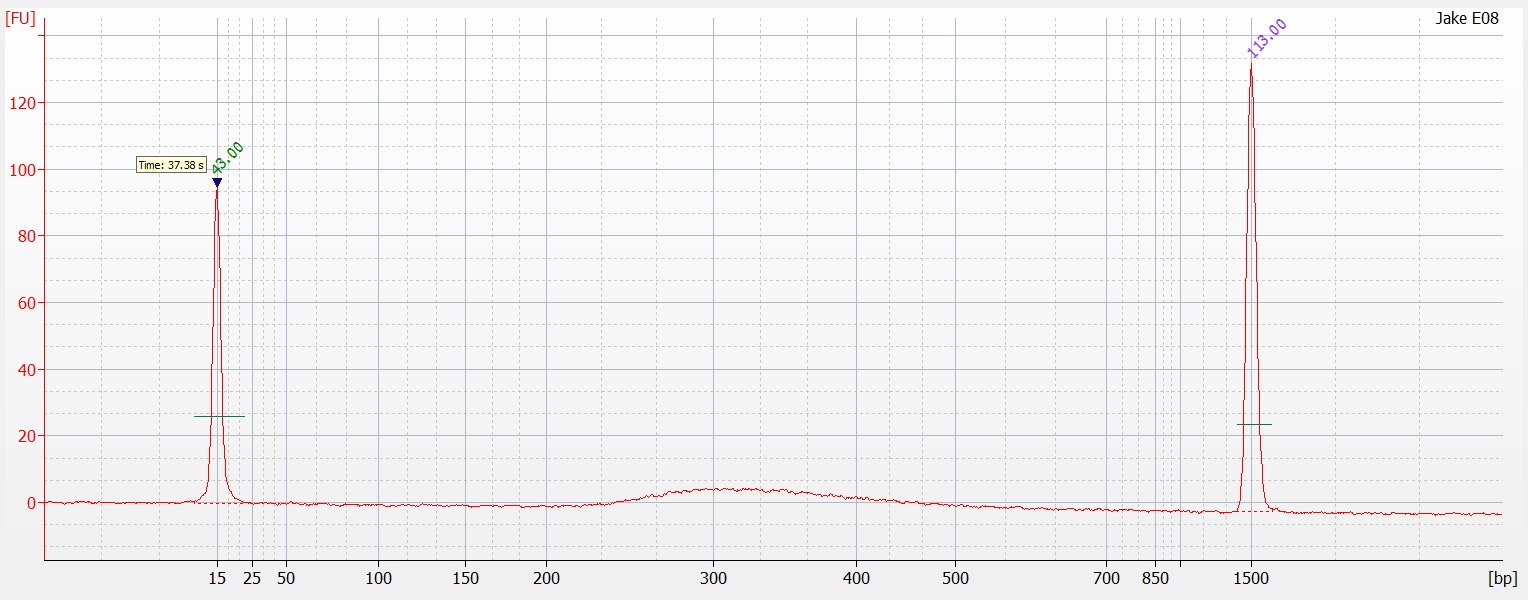
I have no idea where the DNA when. I saw a ton of it yesterday when I was extracting the DNA. I'm wondering if I missed it. When I was pulling the sample to load as it may have sank to the bottom of the tube and I collected off the top. On the bright side you can see two bands of HQ High molecular weight DNA. in the Centrifuged set. I will run another gel tomorrow to see if I can get better samples to appear on here.
This makes me wonder if there is something wrong with the freezer tissue that has degraded the High molecular weight stuff. I talked to Andy yesterday and he told me all of his sablefish samples were stored in EtOH to avoid degradation. Either way this is up for consideration.
**Update 3/13/2015**
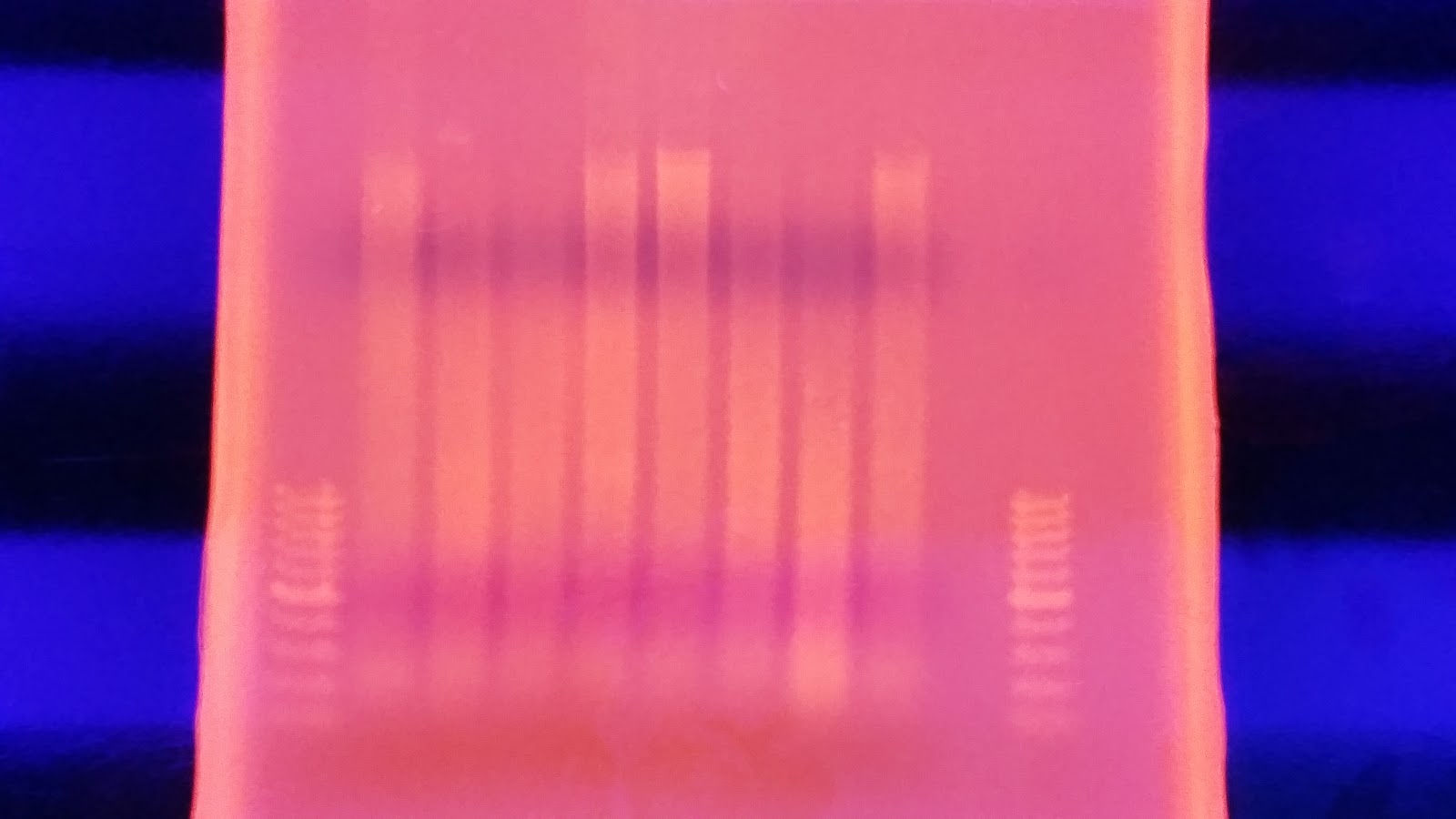
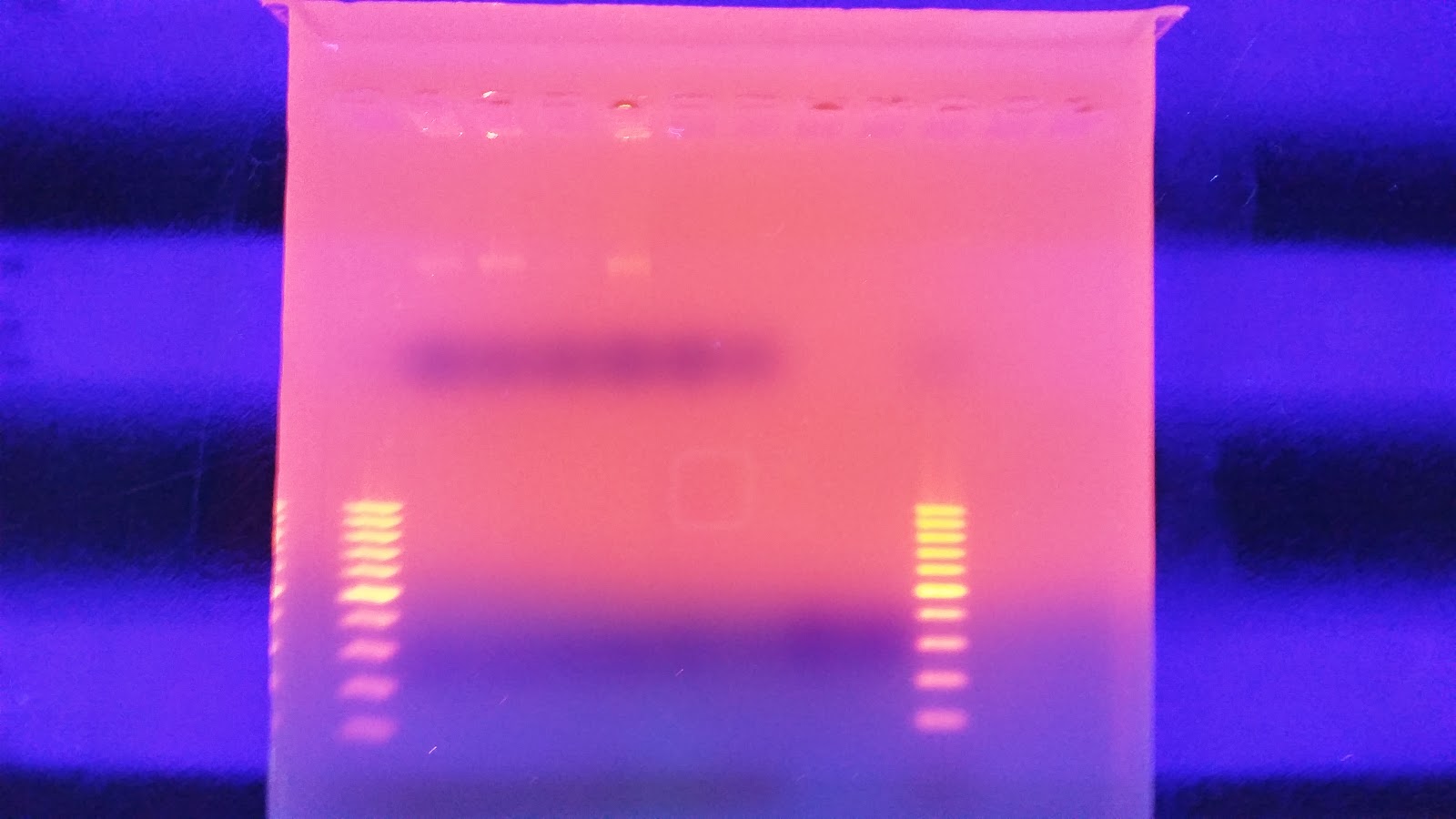
I ran the same protocol again this morning except this time I mixed the samples very well before pulling a sample from them. After running the gel, it looks almost identical except sample #1 looks better in the second sample. I'm not sure what happened with this protocol but supposedly that single band is confirmation of high quality high molecular weight DNA.
**Update 3/13/2015**
I ran the same protocol again this morning except this time I mixed the samples very well before pulling a sample from them. After running the gel, it looks almost identical except sample #1 looks better in the second sample. I'm not sure what happened with this protocol but supposedly that single band is confirmation of high quality high molecular weight DNA.