The qPCR reagent table:
| Volume | Reactions X12 | |
| Ssofast Evagreen MM | 10 | 280 |
| FWD Primer | 0.5 | 14 |
| REV Primer | 0.5 | 14 |
| Nuclease Free H2O | 8.5 | 238 |
| RNA | 0.5 |
For the qPCR I used Actin primers and a positive control from Fidalgo seed oysters extracted on 3/23/2015 with a concentration of 167.3 ng/ul. My no template control contained no DNA as to show there was no contamination in the Master Mix.
qPCR protocol:
1. Added each from greatest volume to least to make the master mix.
2. Pipetted 19.5 ul master mix into each well of a qPCR partial plate
3. Added 0.5 ul sample to each tube
Table Layout.
| 9 | 10 | 11 | 12 |
| C- | C+ | C- | C+ |
| 42715HM1 | 42715SM1 | 42715HT1 | 42715ST1 |
| 42715HM2 | 42715SM2 | 42715HT2 | 42715ST2 |
| 42715HM3 | 42715SM3 | 42715HT3 | 42715ST3 |
| 42715HM4 | 42715SM4 | 42715HT4 | 42715ST4 |
| 42715HM5 | 42715SM5 | 42715HT5 | 42715ST5 |
I ran the following program:
| Sybr New Plate+Sybr cDNA 55 melt 2 | ||
| Step | Temperature | Time |
| Initiation | 95 C | 10 min |
| Elongation | 95 C | 15 sec |
| 55 C | 15 sec | |
| Read | ||
| 72 C | 15 sec | |
| Read | ||
| Repeat Elongation 40 times | ||
| Termination | 95 C | 1 min |
| 55 C | 1 sec | |
| Melt Curve Manual ramp 0.2C per sec Read 0.5 C | 65 - 95 C | 30 sec |
| 21 C | 10 min | |
| End |
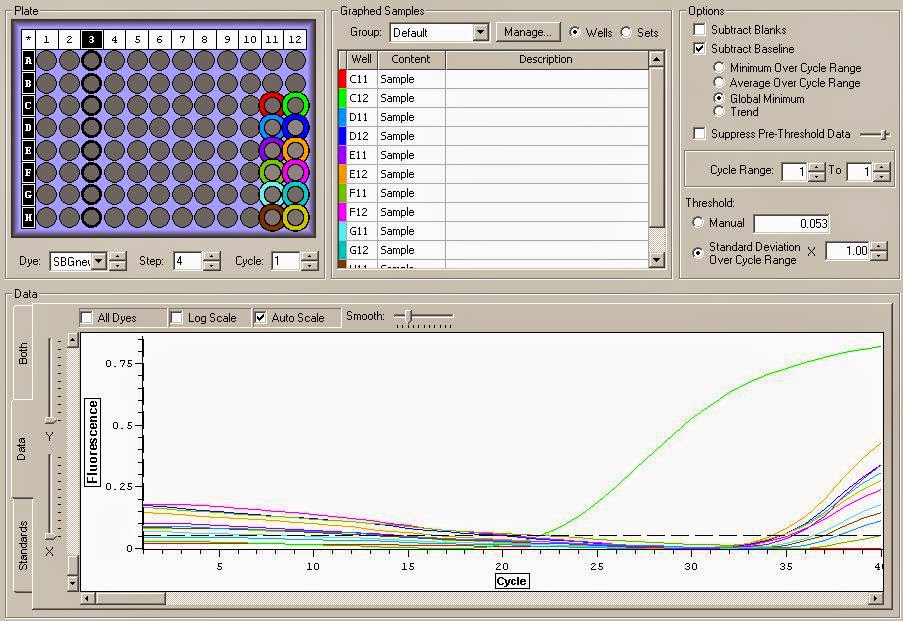
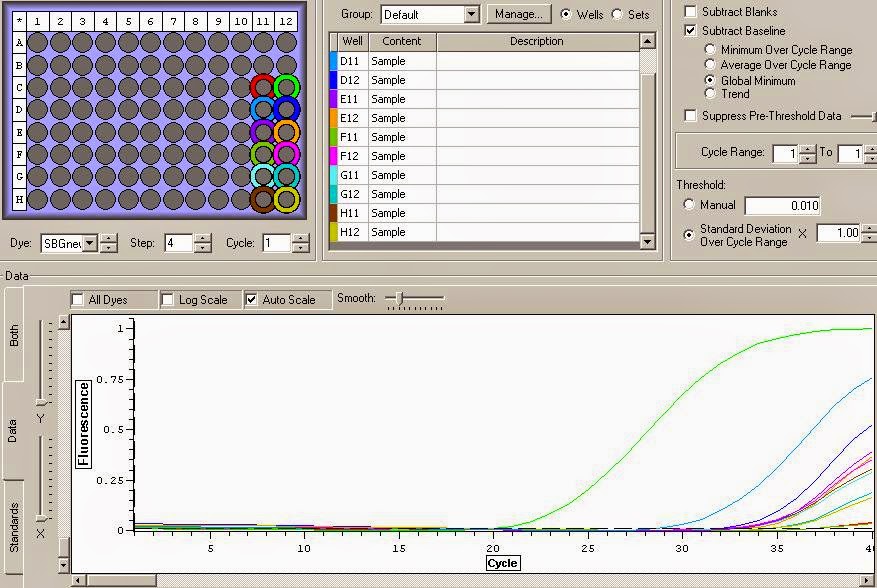
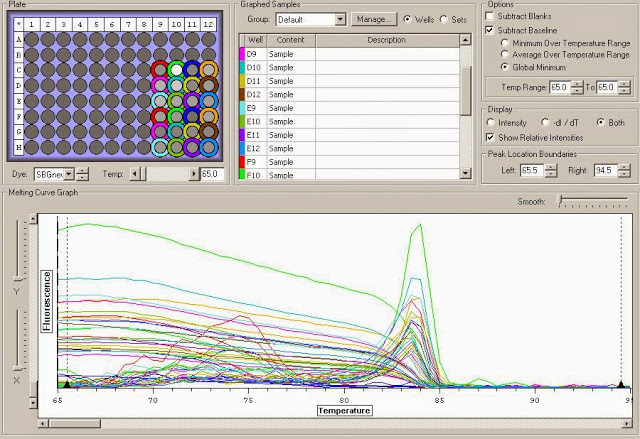
You can see the amplification curves below:
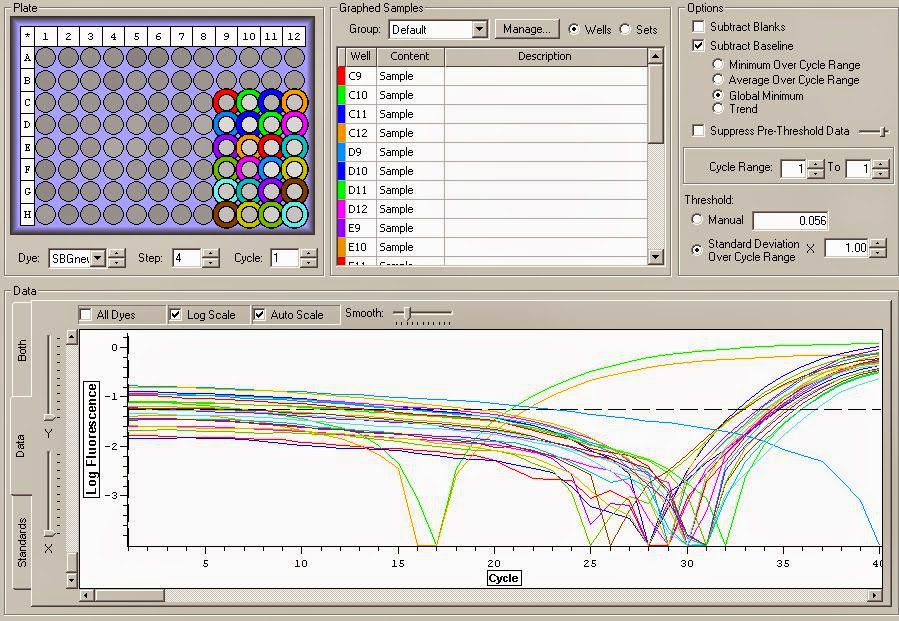
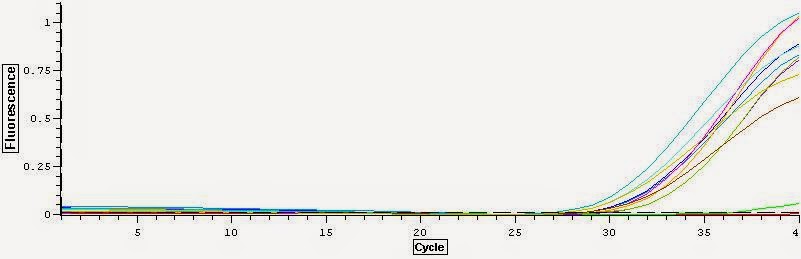
You can see the Melt curve below:
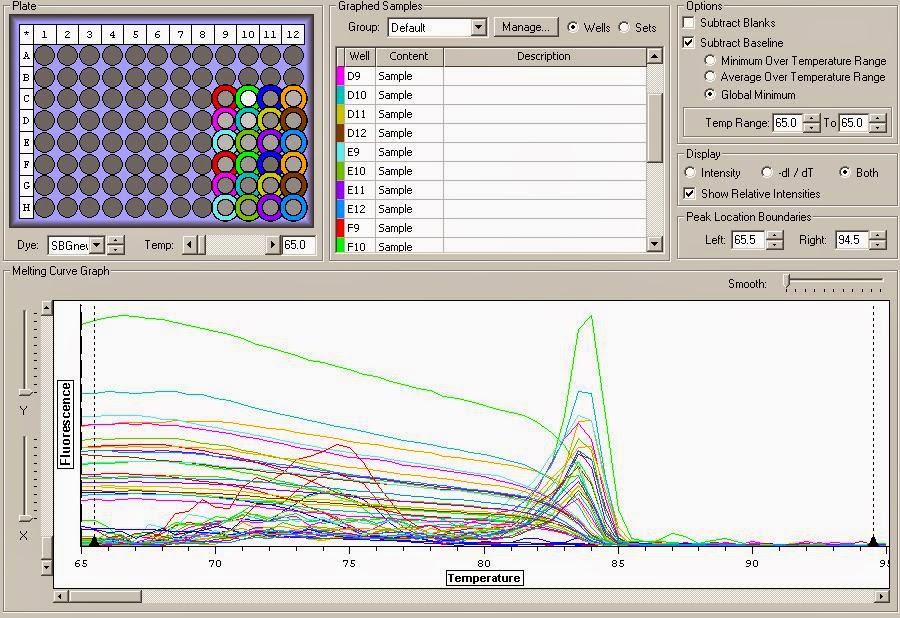
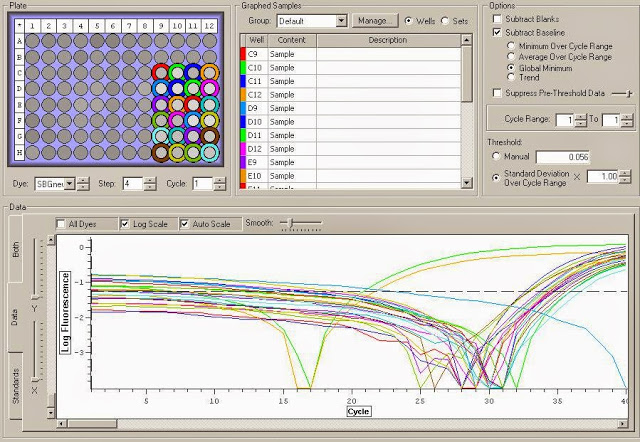
It appears that there is definite gDNA contamination in the samples which is a shame. To remedy this I will be running another round of DNase treatment on them today. I'll check them with another qPCR tonight. You can view the raw qPCR file here.