All runs (3 runs were conducted) were created using a master mix containing C.gigas gDNA (either 50ng or 100ng), 1X Promega qPCR Master Mix, 0.2uM each of forward/reverse primers (18s; Roberts SR ID: 156, 157). The master mix was mixed well and 10uL were distributed in each well of ABI plates. Plates were sealed with ABI optical adhesive covers.
It should also be noted that this analysis was only done with a single primer set and was not tested on any other qPCR machines. This can easily be done if it is desired, however I think one of the issues still being observed with the machine is sample-independent (see Results section below).
Results:
Here’s an extremely quick and dirty analysis of what these qPCR runs have revealed (across the entire plate, 3 plates of data):
Avg. Range of Cts Across Plates – 1.70
Avg. Std. Deviation of Cts Across Plates – 0.352
Based off of the graphs below (particularly the Ct vs Well Position plot), my conclusion is that the machine reads plates inaccurately in Rows A, B, C, F, G, & H. Rows D & E exhibit the most consistent well-to-well readings and, potentially, could be used for qPCR.
The entire work up (which includes a breakdown of each well position relative to each other) is here (Excel Workbook .xlsx). Below are screen captures of one of the three plates (as an example, since all looked the same) that were used for analysis of the amplification plots, melt curves and Ct vs Well Position and a quick description/assessment of what I have observed.
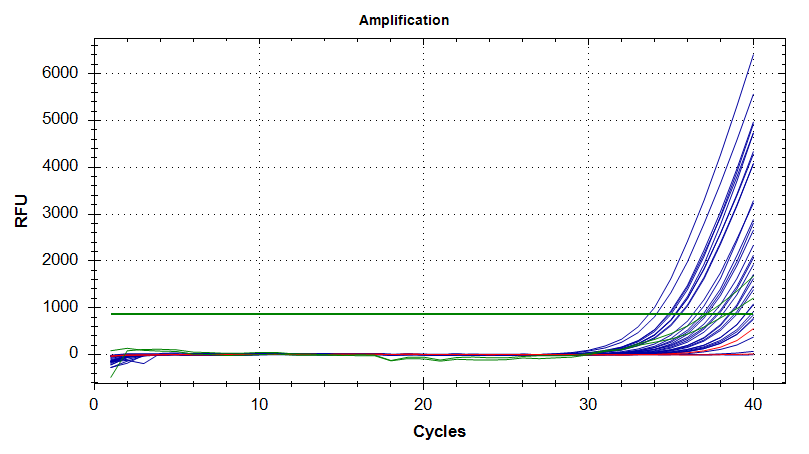
The amplification plot (below) clearly shows the type of spread in Cts across an entire plate that was observed in each run, as well as a large range in fluorescence detected (Rn) in each well.

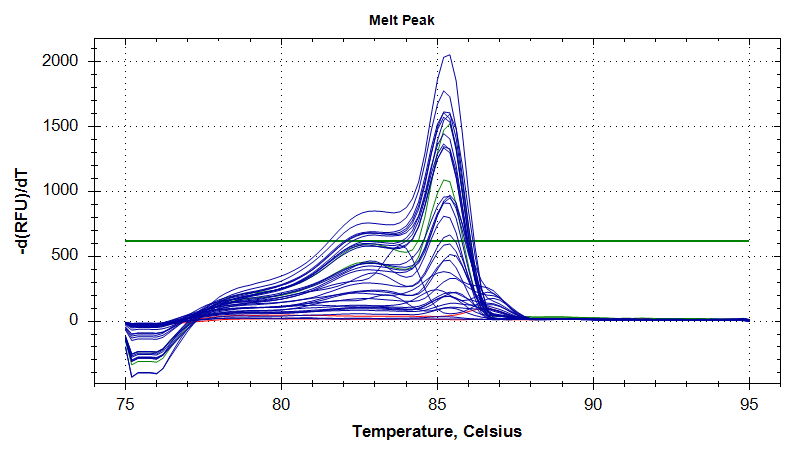
The melt curve (below) reflects the large range of detected fluorescence seen in the amplification plot. Additionally, some wells exhibit small “bumps” between 75C and 80C. This provides more evidence for a problem with well-to-well consistency.

A graph of Ct vs. Well Position (below) reveals some enlightening information. From looking at this plot, it’s clear that the machine reads from A1 to A12, then B1 to B12 (reads by row, not column) and so on. This plot reveals that most of the variation seen in Ct values occurs in the two rows closest to the edge of the plate, and within those rows, the middle wells’ Cts are more similar to the Cts observed throughout the rest of the plate.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}