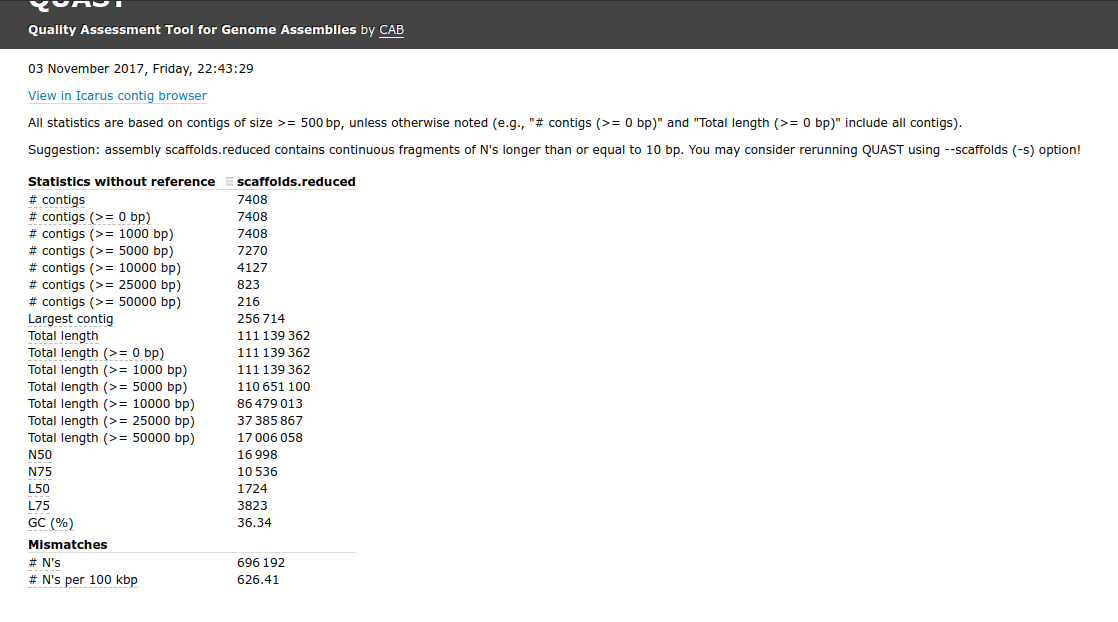
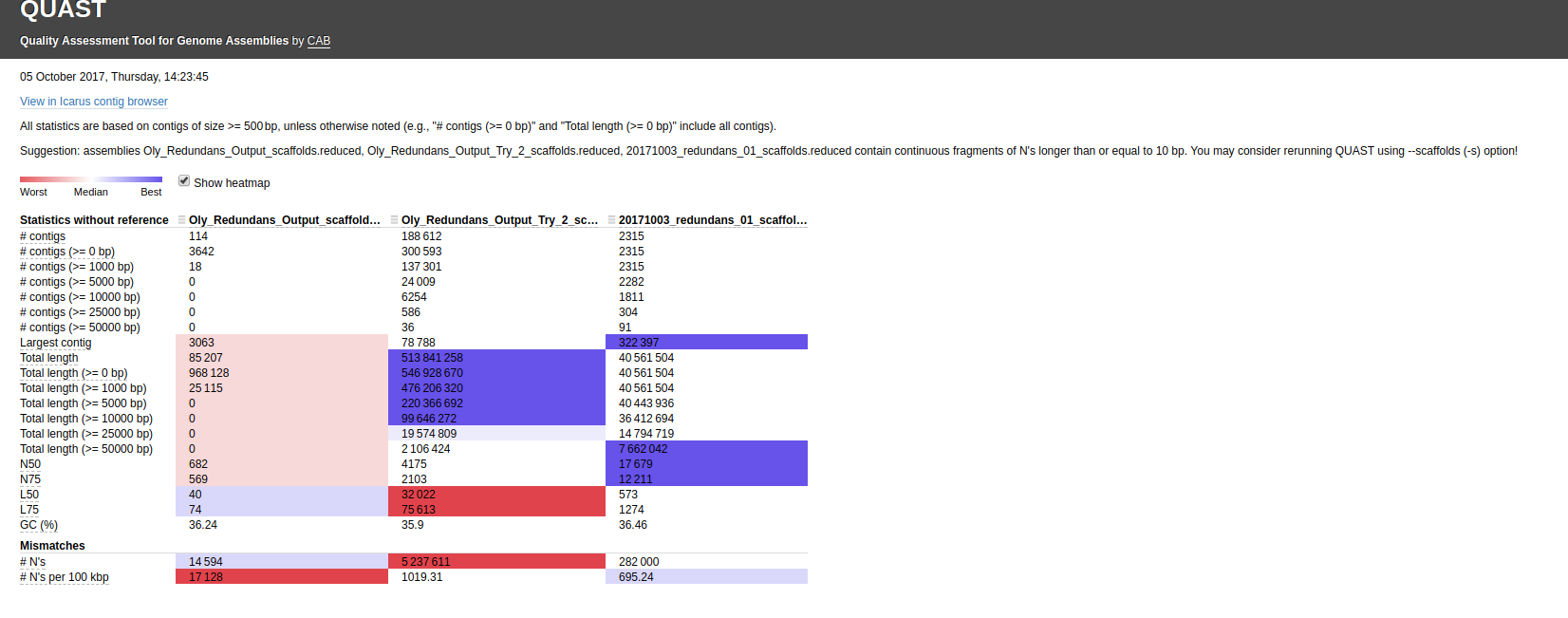
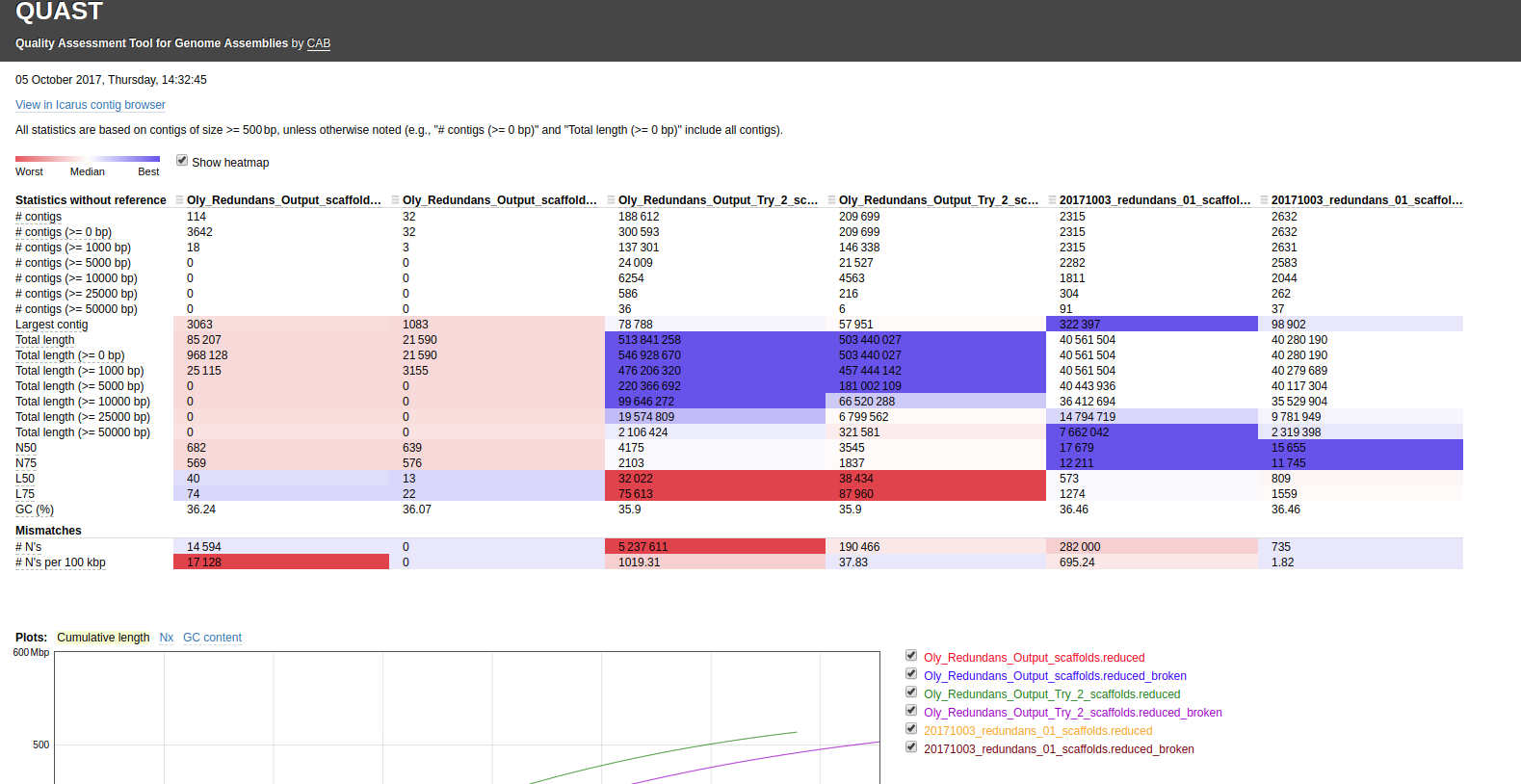
Ran this on Mox (hyak) node.
Create single PacBio FASTA file:
list of PacBio files:
m130619_081336_42134_c100525122550000001823081109281326_s1_p0.fastq m170315_001112_42134_c101169372550000001823273008151717_s1_p0_filtered_subreads.fastq.gz
m170211_224036_42134_c101073082550000001823236402101737_s1_X0_filtered_subreads.fastq.gz m170315_063041_42134_c101169382550000001823273008151700_s1_p0_filtered_subreads.fastq.gz
m170301_100013_42134_c101174162550000001823269408211761_s1_p0_filtered_subreads.fastq.gz m170315_124938_42134_c101169382550000001823273008151701_s1_p0_filtered_subreads.fastq.gz
m170301_162825_42134_c101174162550000001823269408211762_s1_p0_filtered_subreads.fastq.gz m170315_190851_42134_c101169382550000001823273008151702_s1_p0_filtered_subreads.fastq.gz
m170301_225711_42134_c101174162550000001823269408211763_s1_p0_filtered_subreads.fastq.gz
m170308_163922_42134_c101174252550000001823269408211742_s1_p0_filtered_subreads.fastq.gz
m170308_230815_42134_c101174252550000001823269408211743_s1_p0_filtered_subreads.fastq.gz
Concatenate GZIPPED files:
$cat *.gz > pacbio_cat.fastq.gz
Convert FASTQ gzip to FASTA:
$zcat pacbio_cat.fastq.gz | awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' > oly_pacbio_concatentated.fa
Convert and concatenate single non-gzipped FASTQ file:
$awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' m130619_081336_42134_c100525122550000001823081109281326_s1_p0.fastq >> oly_pacbio_concatentated.fa
$cat *.gz > pacbio_cat.fastq.gz
GUNZIP Illumina GZIPPED FASTQ
$for i in *.gz; do gunzip < "$i" > ${i%%.gz}; done
Determine mean read length and standard deviation from Illumina FASTQ files (needed for MaSuRCA config file) (found code below from this Biostars thread:
$for i in *.fq; do echo "$i "; awk 'BEGIN { t=0.0;sq=0.0; n=0;} ;NR%4==2 {n++;L=length($0);t+=L;sq+=L*L;}END{m=t/n;printf("total %d avg=%f stddev=%f\n",n,m,sq/n-m*m);}' $i; done
Output:
151114_I191_FCH3Y35BCXX_L1_wHAIPI023992-37_1.fq
total 61253141 avg=150.000000 stddev=0.000000
151114_I191_FCH3Y35BCXX_L1_wHAIPI023992-37_2.fq
total 61253141 avg=150.000000 stddev=0.000000
151114_I191_FCH3Y35BCXX_L2_wHAMPI023991-66_1.fq
total 58755925 avg=150.000000 stddev=0.000000
151114_I191_FCH3Y35BCXX_L2_wHAMPI023991-66_2.fq
total 58755925 avg=150.000000 stddev=0.000000
151118_I137_FCH3KNJBBXX_L5_wHAXPI023905-96_1.fq
total 43938762 avg=150.000000 stddev=0.000000
151118_I137_FCH3KNJBBXX_L5_wHAXPI023905-96_2.fq
total 43938762 avg=150.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCABDLAAPEI-62_1.fq
total 87198584 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCABDLAAPEI-62_2.fq
total 87198584 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCACDTAAPEI-75_1.fq
total 43766527 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCACDTAAPEI-75_2.fq
total 43766527 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCABDLAAPEI-62_1.fq
total 87135961 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCABDLAAPEI-62_2.fq
total 87135961 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCACDTAAPEI-75_1.fq
total 43138844 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCACDTAAPEI-75_2.fq
total 43138844 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L5_WHOSTibkDCAADWAAPEI-74_1.fq
total 95270180 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L5_WHOSTibkDCAADWAAPEI-74_2.fq
total 95270180 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L6_WHOSTibkDCAADWAAPEI-74_1.fq
total 92524416 avg=49.000000 stddev=0.000000
160103_I137_FCH3V5YBBXX_L6_WHOSTibkDCAADWAAPEI-74_2.fq
total 92524416 avg=49.000000 stddev=0.000000
Here’s the config file I’m using:
/gscratch/scrubbed/samwhite/20171019_masurca_oly_assembly/20171019_masurca_oly_config.txt
# example configuration file
# DATA is specified as type {PE,JUMP,OTHER,PACBIO} and 5 fields:
# 1)two_letter_prefix 2)mean 3)stdev 4)fastq(.gz)_fwd_reads
# 5)fastq(.gz)_rev_reads. The PE reads are always assumed to be
# innies, i.e. --->.<---, and JUMP are assumed to be outties
# <---.--->. If there are any jump libraries that are innies, such as
# longjump, specify them as JUMP and specify NEGATIVE mean. Reverse reads
# are optional for PE libraries and mandatory for JUMP libraries. Any
# OTHER sequence data (454, Sanger, Ion torrent, etc) must be first
# converted into Celera Assembler compatible .frg files (see
# http://wgs-assembler.sourceforge.com)
DATA
PE= pe 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCABDLAAPEI-62_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCABDLAAPEI-62_2.fq
PE= pe 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCACDTAAPEI-75_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCACDTAAPEI-75_2.fq
PE= pe 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCABDLAAPEI-62_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCABDLAAPEI-62_2.fq
PE= pe 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCACDTAAPEI-75_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCACDTAAPEI-75_2.fq
PE= pe 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L5_WHOSTibkDCAADWAAPEI-74_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L5_WHOSTibkDCAADWAAPEI-74_2.fq
PE= pe 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L6_WHOSTibkDCAADWAAPEI-74_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L6_WHOSTibkDCAADWAAPEI-74_2.fq
JUMP= sh 150 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L1_wHAIPI023992-37_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L1_wHAIPI023992-37_2.fq
JUMP= sh 150 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L2_wHAMPI023991-66_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L2_wHAMPI023991-66_2.fq
JUMP= sh 150 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151118_I137_FCH3KNJBBXX_L5_wHAXPI023905-96_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151118_I137_FCH3KNJBBXX_L5_wHAXPI023905-96_2.fq
#pacbio reads must be in a single fasta file! make sure you provide absolute path
PACBIO=/gscratch/scrubbed/samwhite/O_lurida/oly_pacbio/oly_pacbio_concatentated.fa
END
PARAMETERS
#this is k-mer size for deBruijn graph values between 25 and 127 are supported, auto will compute the optimal size based on the read data and GC content
GRAPH_KMER_SIZE = auto
#set this to 1 for all Illumina-only assemblies
#set this to 1 if you have less than 20x long reads (454, Sanger, Pacbio) and less than 50x CLONE coverage by Illumina, Sanger or 454 mate pairs
#otherwise keep at 0
USE_LINKING_MATES = 1
#this parameter is useful if you have too many Illumina jumping library mates. Typically set it to 60 for bacteria and 300 for the other organisms
LIMIT_JUMP_COVERAGE = 300
#these are the additional parameters to Celera Assembler. do not worry about performance, number or processors or batch sizes -- these are computed automatically.
#set cgwErrorRate=0.25 for bacteria and 0.1<=cgwErrorRate<=0.15 for other organisms.
CA_PARAMETERS = cgwErrorRate=0.15
#minimum count k-mers used in error correction 1 means all k-mers are used. one can increase to 2 if Illumina coverage >100
KMER_COUNT_THRESHOLD = 1
#whether to attempt to close gaps in scaffolds with Illumina data
CLOSE_GAPS=1
#auto-detected number of cpus to use
NUM_THREADS = 28
#this is mandatory jellyfish hash size -- a safe value is estimated_genome_size*estimated_coverage
JF_SIZE = 200000000
#set this to 1 to use SOAPdenovo contigging/scaffolding module. Assembly will be worse but will run faster. Useful for very large (>5Gbp) genomes
SOAP_ASSEMBLY=0
END
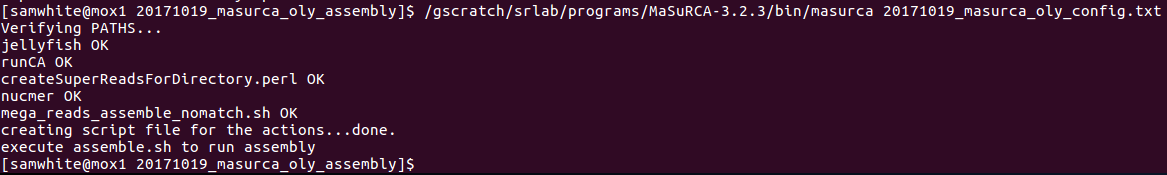
Execute the masurca script to generate assembly script (on Mox login node):
$cd /gscratch/scrubbed/samwhite/20171019_masurca_oly_assembly
$/gscratch/srlab/programs/MaSuRCA-3.2.3/bin/masurca 20171019_masurca_oly_config.txt
Got this error:

I’ll edit config file to have standard deviations == 1 and try again.
Got another error:
I’ll edit config file to have different two letter prefix assignments…
It worked!
Final version of config:
# example configuration file
# DATA is specified as type {PE,JUMP,OTHER,PACBIO} and 5 fields:
# 1)two_letter_prefix 2)mean 3)stdev 4)fastq(.gz)_fwd_reads
# 5)fastq(.gz)_rev_reads. The PE reads are always assumed to be
# innies, i.e. --->.<---, and JUMP are assumed to be outties
# <---.--->. If there are any jump libraries that are innies, such as
# longjump, specify them as JUMP and specify NEGATIVE mean. Reverse reads
# are optional for PE libraries and mandatory for JUMP libraries. Any
# OTHER sequence data (454, Sanger, Ion torrent, etc) must be first
# converted into Celera Assembler compatible .frg files (see
# http://wgs-assembler.sourceforge.com)
DATA
PE= aa 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCABDLAAPEI-62_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCABDLAAPEI-62_2.fq
PE= ab 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCACDTAAPEI-75_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L3_WHOSTibkDCACDTAAPEI-75_2.fq
PE= ac 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCABDLAAPEI-62_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCABDLAAPEI-62_2.fq
PE= ad 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCACDTAAPEI-75_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L4_WHOSTibkDCACDTAAPEI-75_2.fq
PE= ae 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L5_WHOSTibkDCAADWAAPEI-74_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L5_WHOSTibkDCAADWAAPEI-74_2.fq
PE= af 49 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L6_WHOSTibkDCAADWAAPEI-74_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/160103_I137_FCH3V5YBBXX_L6_WHOSTibkDCAADWAAPEI-74_2.fq
JUMP= ba 150 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L1_wHAIPI023992-37_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L1_wHAIPI023992-37_2.fq
JUMP= bb 150 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L2_wHAMPI023991-66_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151114_I191_FCH3Y35BCXX_L2_wHAMPI023991-66_2.fq
JUMP= bc 150 1 /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151118_I137_FCH3KNJBBXX_L5_wHAXPI023905-96_1.fq /gscratch/scrubbed/samwhite/O_lurida/oly_illumina/151118_I137_FCH3KNJBBXX_L5_wHAXPI023905-96_2.fq
#pacbio reads must be in a single fasta file! make sure you provide absolute path
PACBIO=/gscratch/scrubbed/samwhite/O_lurida/oly_pacbio/oly_pacbio_concatentated.fa
END
PARAMETERS
#this is k-mer size for deBruijn graph values between 25 and 127 are supported, auto will compute the optimal size based on the read data and GC content
GRAPH_KMER_SIZE = auto
#set this to 1 for all Illumina-only assemblies
#set this to 1 if you have less than 20x long reads (454, Sanger, Pacbio) and less than 50x CLONE coverage by Illumina, Sanger or 454 mate pairs
#otherwise keep at 0
USE_LINKING_MATES = 1
#this parameter is useful if you have too many Illumina jumping library mates. Typically set it to 60 for bacteria and 300 for the other organisms
LIMIT_JUMP_COVERAGE = 300
#these are the additional parameters to Celera Assembler. do not worry about performance, number or processors or batch sizes -- these are computed automatically.
#set cgwErrorRate=0.25 for bacteria and 0.1<=cgwErrorRate<=0.15 for other organisms.
CA_PARAMETERS = cgwErrorRate=0.15
#minimum count k-mers used in error correction 1 means all k-mers are used. one can increase to 2 if Illumina coverage >100
KMER_COUNT_THRESHOLD = 1
#whether to attempt to close gaps in scaffolds with Illumina data
CLOSE_GAPS=1
#auto-detected number of cpus to use
NUM_THREADS = 28
#this is mandatory jellyfish hash size -- a safe value is estimated_genome_size*estimated_coverage
JF_SIZE = 200000000
#set this to 1 to use SOAPdenovo contigging/scaffolding module. Assembly will be worse but will run faster. Useful for very large (>5Gbp) genomes
SOAP_ASSEMBLY=0
END
Submitted the job to Mox using the following command:
sbatch -p srlab -A srlab 20171019_masurca_oly_assembly.sh
Here’s the sbatch script used for the job:
#!/bin/bash
## Job Name
#SBATCH --job-name=20171019_masurca_oly
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes (We only get 1, so this is fixed)
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=30-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/20171019_masurca_oly_assembly
/gscratch/scrubbed/samwhite/20171019_masurca_oly_assembly/assemble.sh