After receiving our RNAseq data from Genewiz earlier today, needed to run FastQC, trim, check trimmed reads with FastQC.
FastQC on raw reads was run locally and files were kept on owl/nightingales/C_bairdi.
fastp trimming was run on Mox, followed by MultiQC.
FastQC on trimmed reads were run locally, followed by MultiQC.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=cbai_fastp_trimming_RNAseq
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=10-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20200318_cbai_RNAseq_fastp_trimming
### C.bairdi RNAseq trimming using fastp.
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
# Set number of CPUs to use
threads=27
# Input/output files
trimmed_checksums=trimmed_fastq_checksums.md5
raw_reads_dir=/gscratch/scrubbed/samwhite/data/C_bairdi/RNAseq/
# Paths to programs
fastp=/gscratch/srlab/programs/fastp-0.20.0/fastp
multiqc=/gscratch/srlab/programs/anaconda3/bin/multiqc
## Inititalize arrays
fastq_array_R1=()
fastq_array_R2=()
programs_array=()
R1_names_array=()
R2_names_array=()
# Programs array
programs_array=("${fastp}" "${multiqc}")
# Capture program options
for program in "${!programs_array[@]}"
do
{
echo "Program options for ${programs_array[program]}: "
echo ""
${programs_array[program]} -h
echo ""
echo ""
echo "----------------------------------------------"
echo ""
echo ""
} &>> program_options.log || true
done
# Sync raw FastQ files to working directory
rsync --archive --verbose \
"${raw_reads_dir}"*.gz .
# Create array of fastq R1 files
for fastq in *R1*.gz
do
fastq_array_R1+=("${fastq}")
done
# Create array of fastq R2 files
for fastq in *R2*.gz
do
fastq_array_R2+=("${fastq}")
done
# Create array of sample names
## Uses awk to parse out sample name from filename
for R1_fastq in *R1*.gz
do
R1_names_array+=($(echo "${R1_fastq}" | awk -F"." '{print $1}'))
done
# Create array of sample names
## Uses awk to parse out sample name from filename
for R2_fastq in *R2*.gz
do
R2_names_array+=($(echo "${R2_fastq}" | awk -F"." '{print $1}'))
done
# Create list of fastq files used in analysis
for fastq in *.gz
do
echo "${fastq}" >> fastq.list.txt
done
# Run fastp on files
for index in "${!fastq_array_R1[@]}"
do
timestamp=$(date +%Y%m%d%M%S)
R1_sample_name=$(echo "${R1_names_array[index]}")
R2_sample_name=$(echo "${R2_names_array[index]}")
${fastp} \
--in1 "${fastq_array_R1[index]}" \
--in2 "${fastq_array_R2[index]}" \
--detect_adapter_for_pe \
--thread ${threads} \
--html "${R1_sample_name}".fastp-trim."${timestamp}".report.html \
--json "${R1_sample_name}".fastp-trim."${timestamp}".report.json \
--out1 "${R1_sample_name}".fastp-trim."${timestamp}".fq.gz \
--out2 "${R2_sample_name}".fastp-trim."${timestamp}".fq.gz
# Generate md5 checksums for newly trimmed files
{
md5sum "${R1_sample_name}".fastp-trim."${timestamp}".fq.gz
md5sum "${R2_sample_name}".fastp-trim."${timestamp}".fq.gz
} >> "${trimmed_checksums}"
# Remove original FastQ files
rm "${fastq_array_R1[index]}" "${fastq_array_R2[index]}"
done
# Run MultiQC
${multiqc} .
RESULTS
Run time was just under three hours:

NOTE: Although the job indicates “FAILED”, this was simply due to a MultiQC failing (path to MultiQC was incorrect). Trimming proceeded/completed properly.
Output folder:
fastp MultiQC report (HTML):
Individual fastp reports are also available (HTML). An example is below.
https://gannet.fish.washington.edu/Atumefaciens/20200318_cbai_RNAseq_fastp_trimming/485_R1_001.fastp-trim.202003181245_fastqc.html
FastQC MultiQC report (HTML):
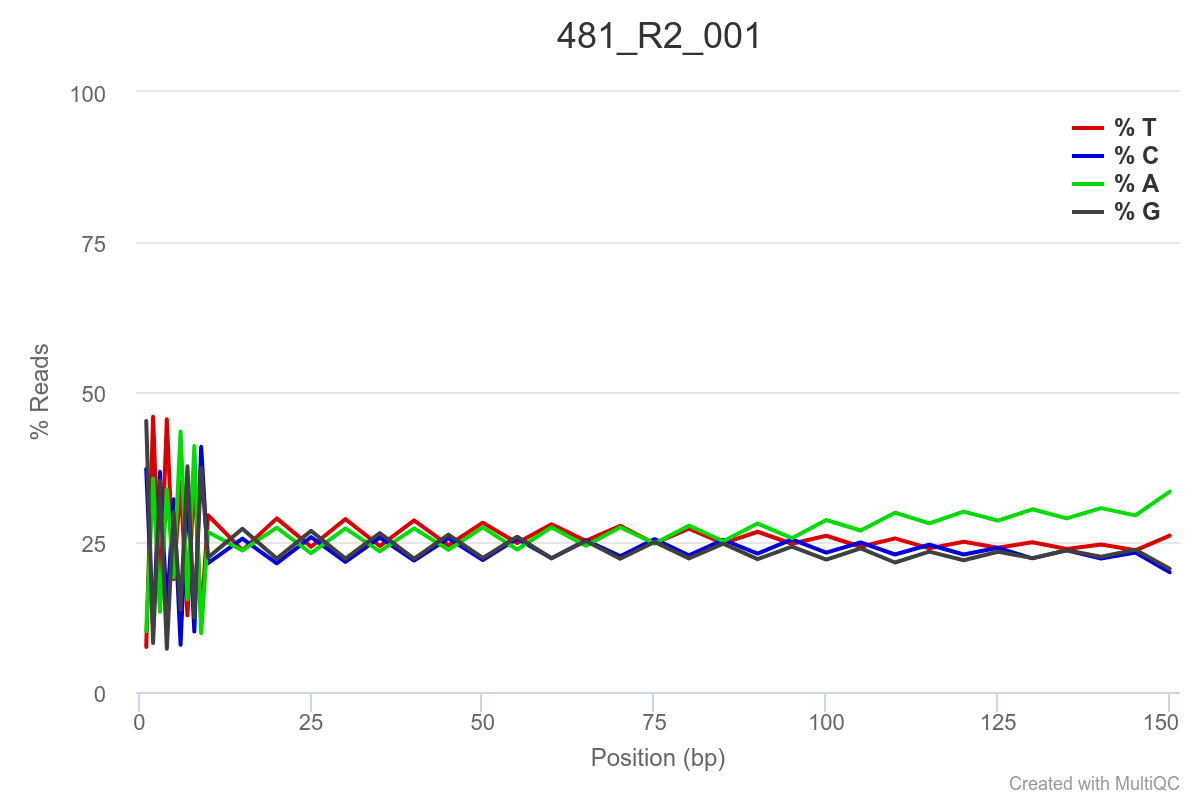
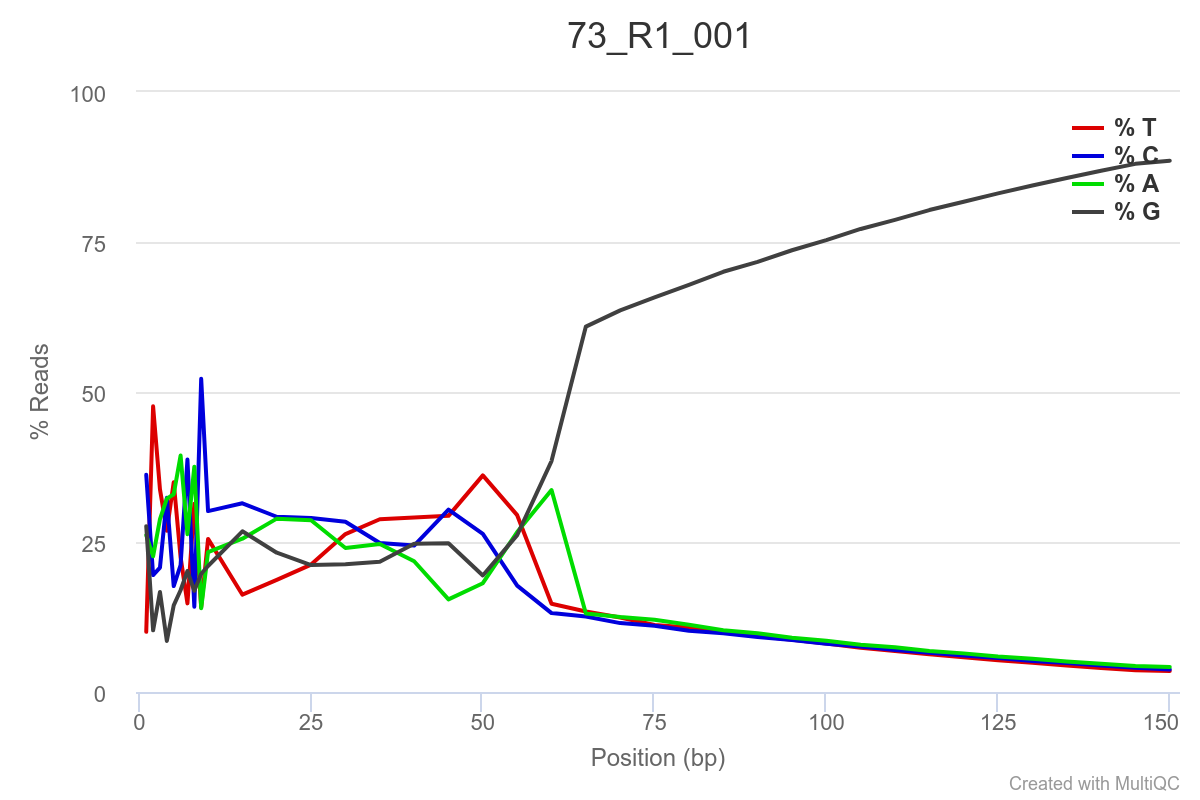
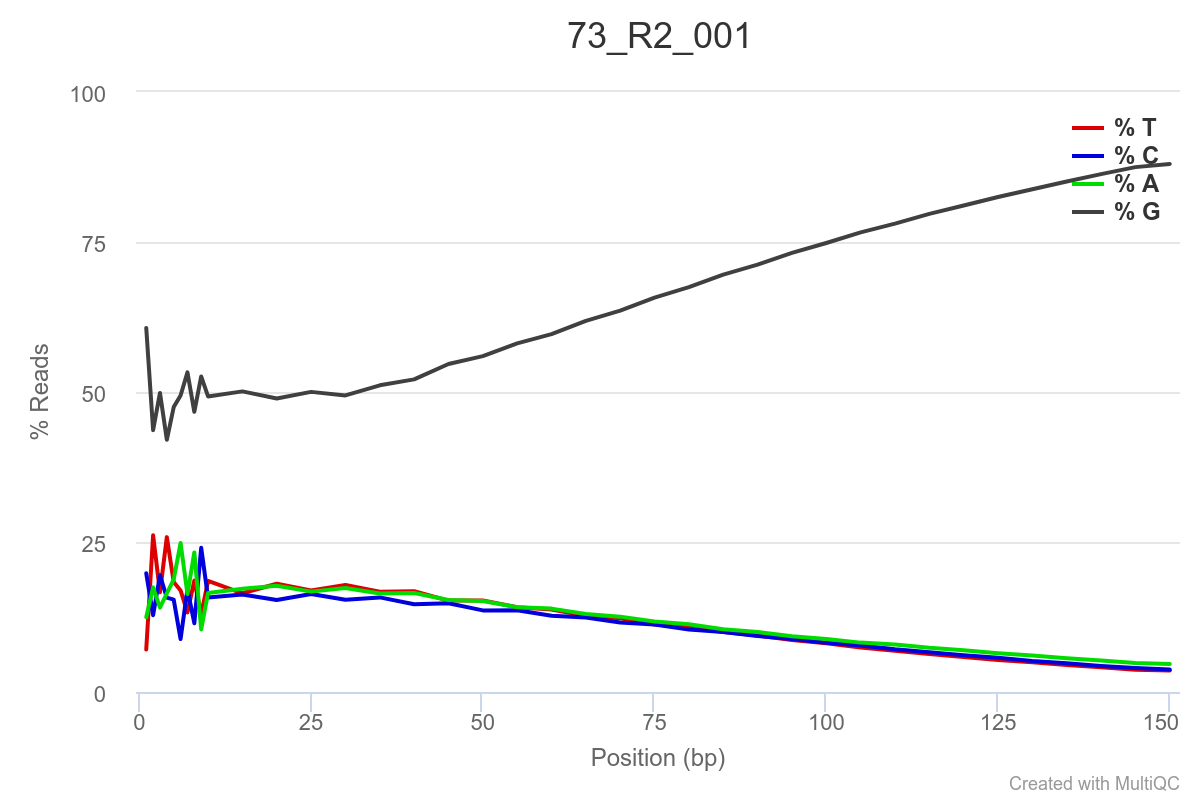
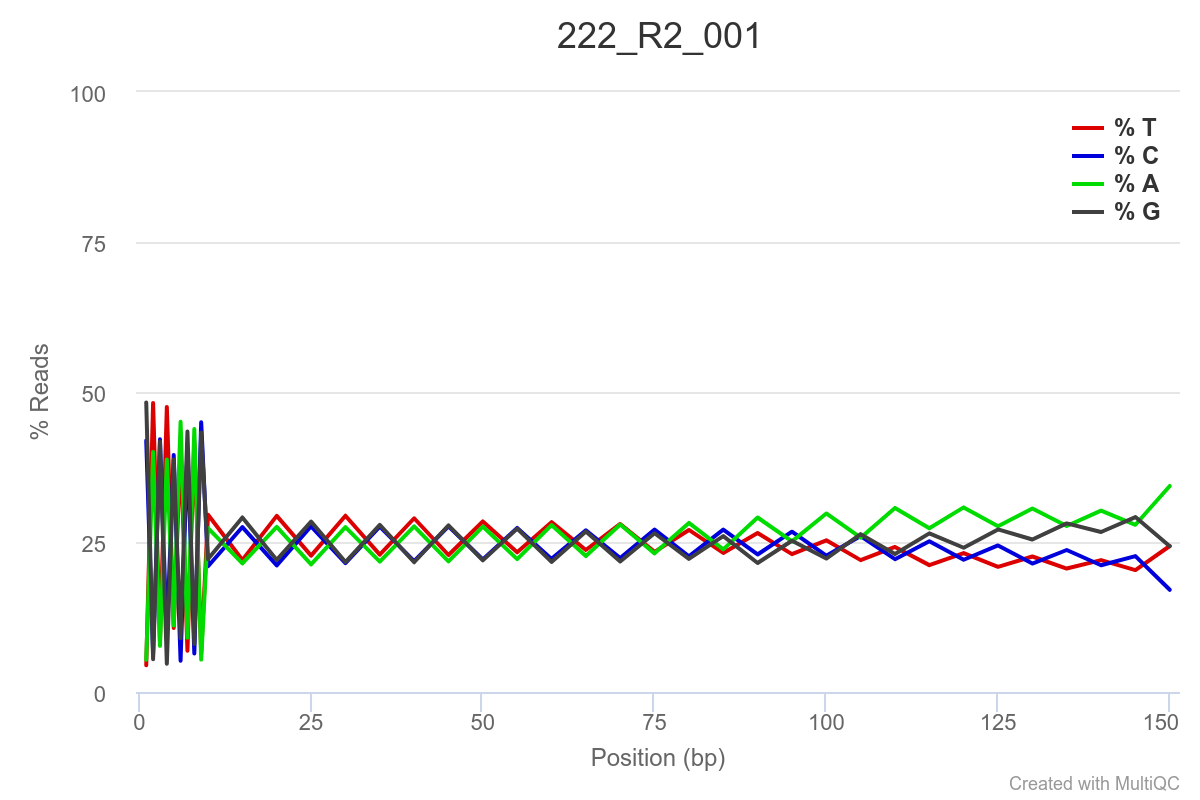
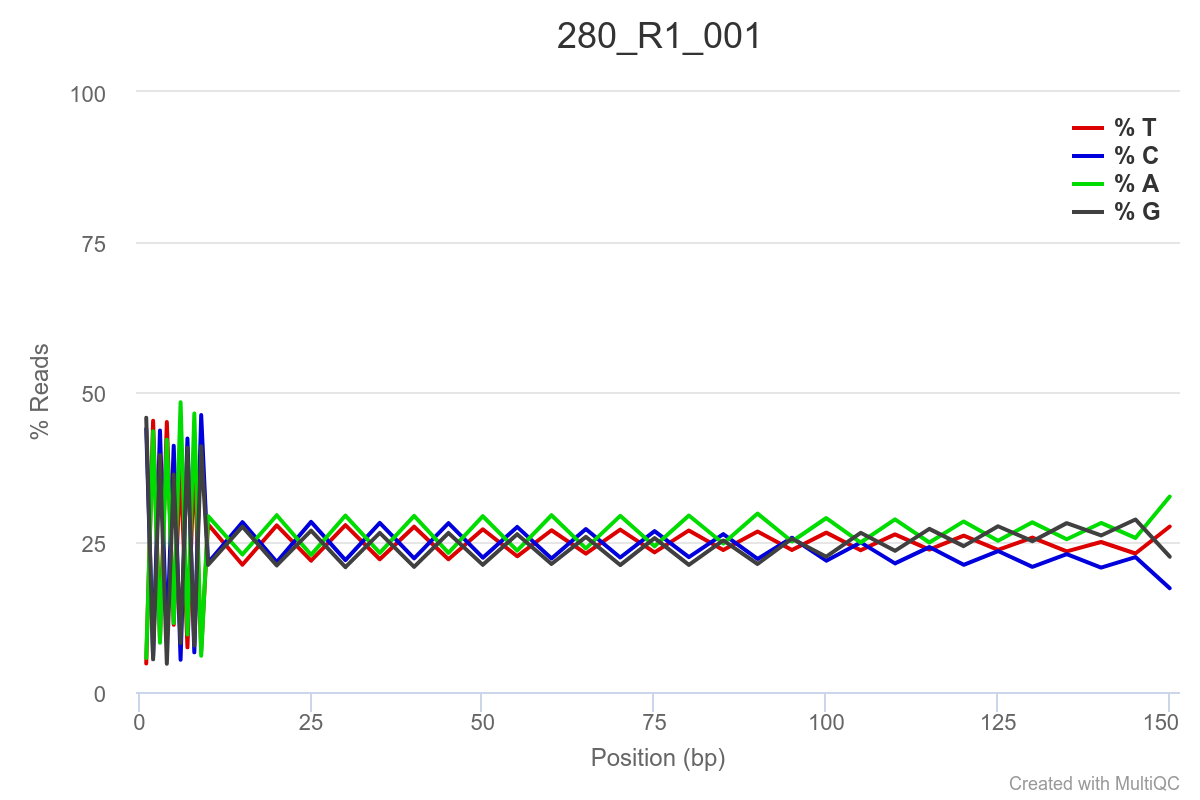
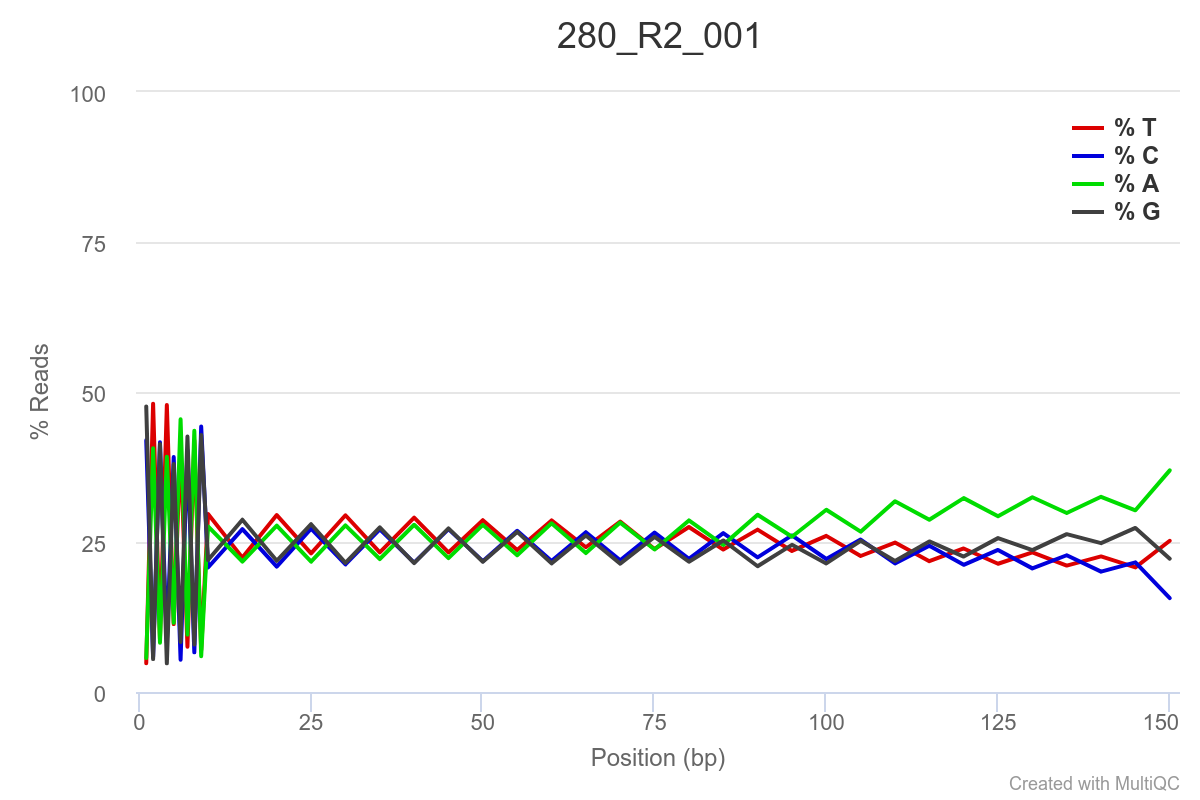
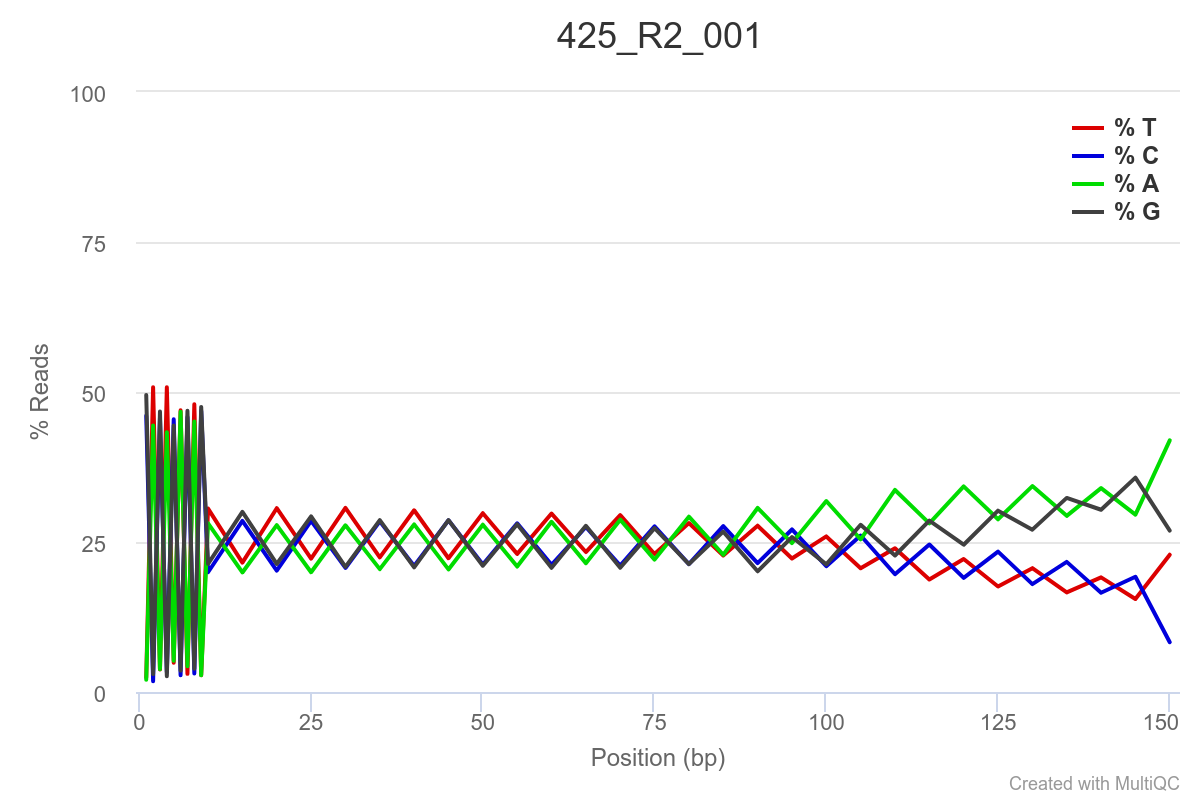
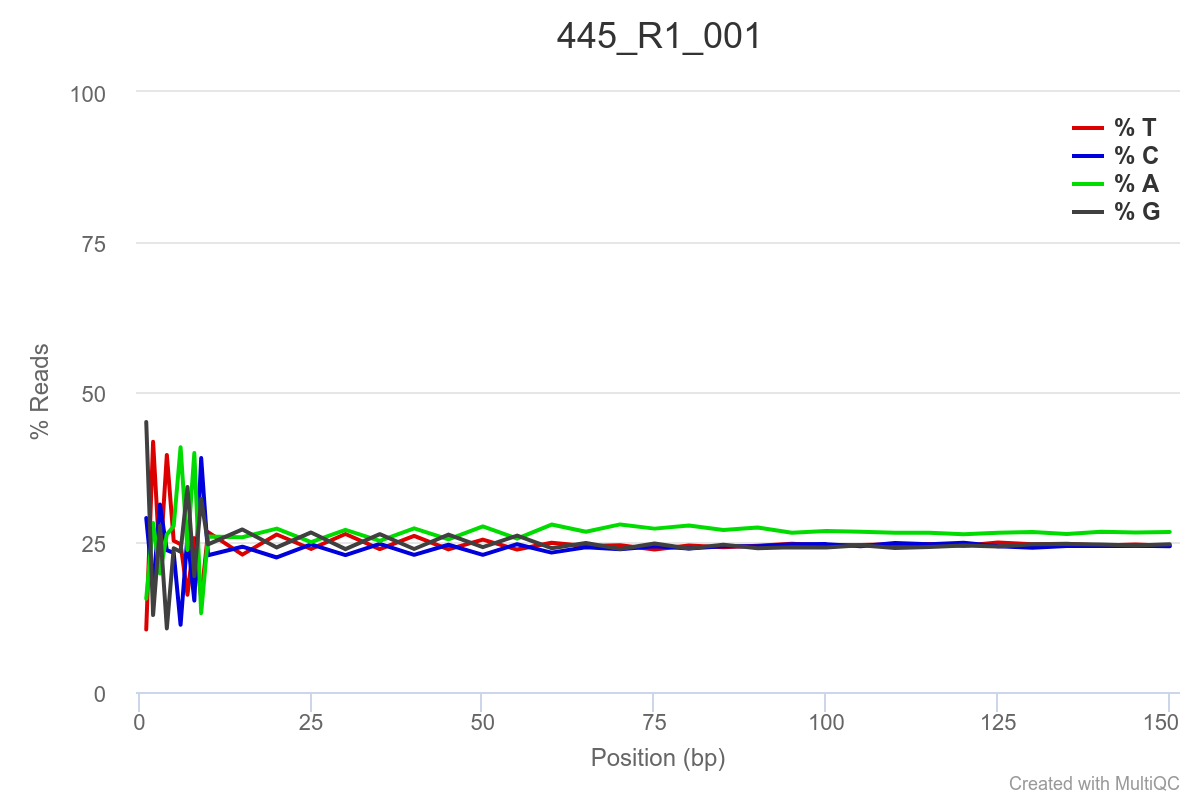
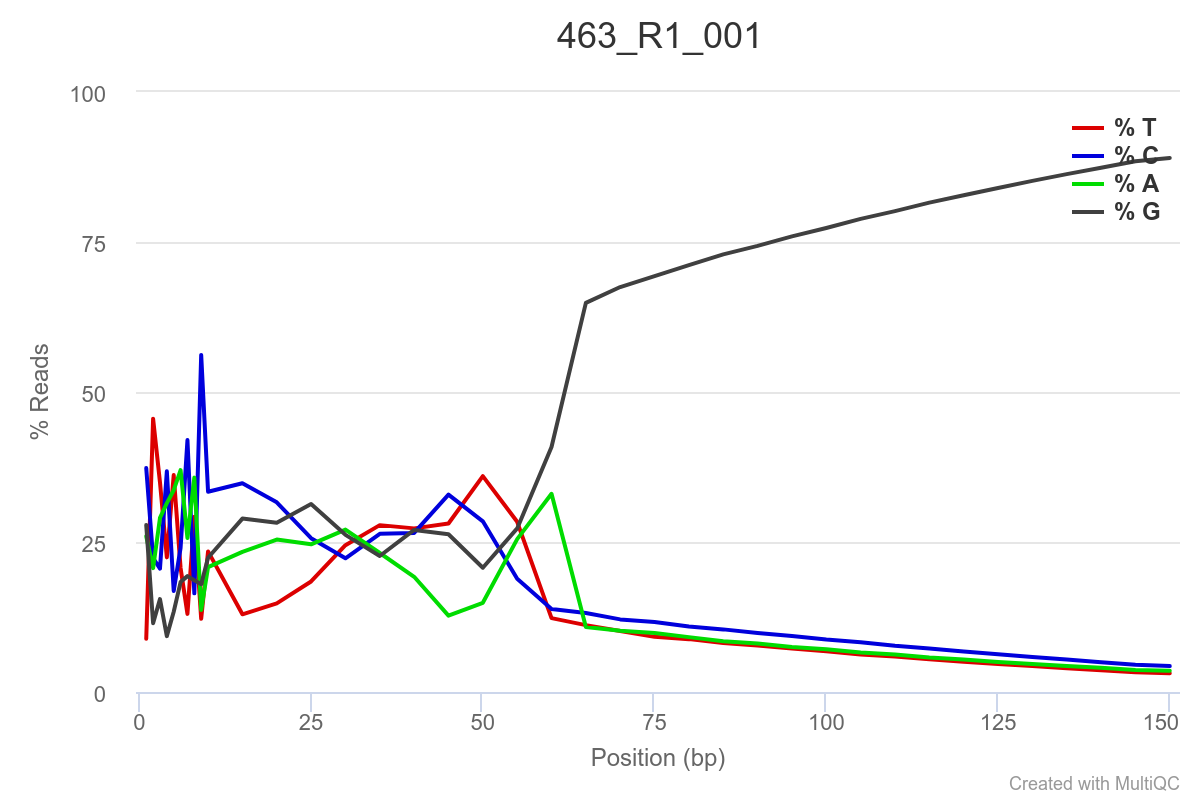
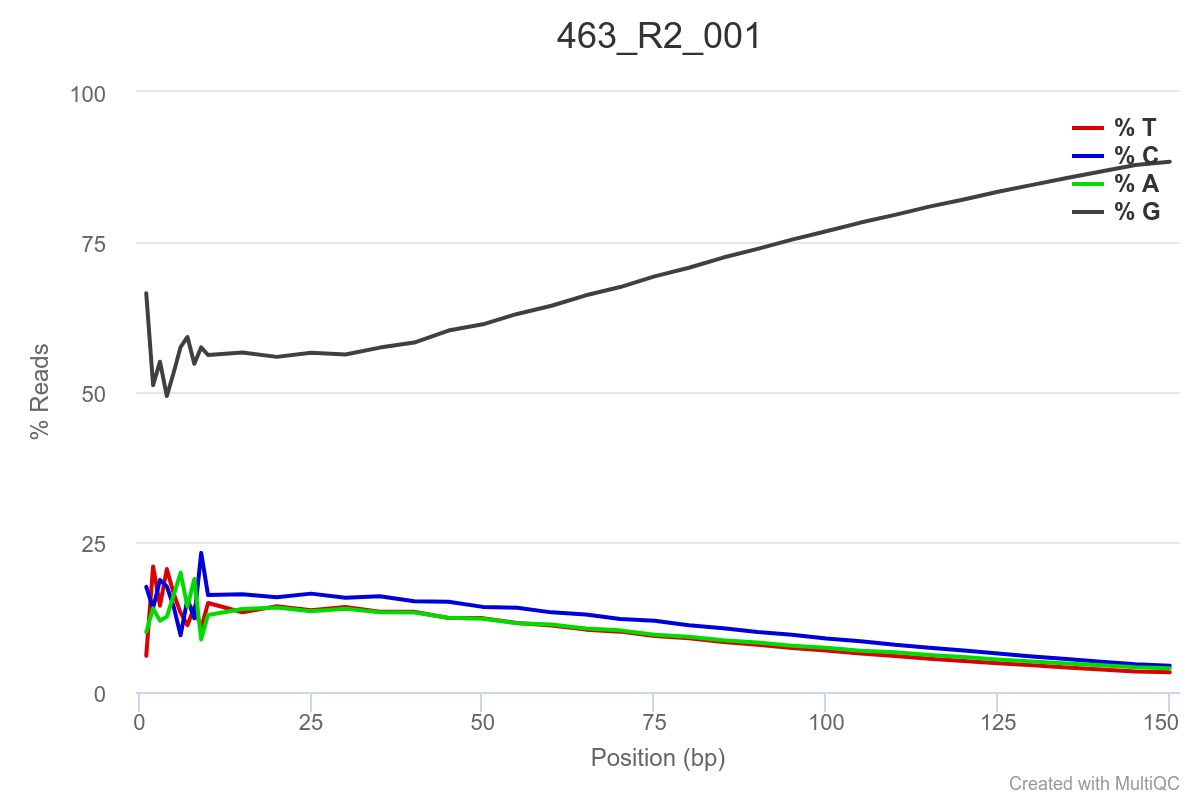
Some of the samples are potentially problematic, based on FastQC plots (see end of post). Despite the weirdness, I think I’m going to leave things as they are and try to filter these reads out downstream. Downstream stuff entails:
-
BLASTx
-
taxonomic read assignment using MEGAN6
I feel like crappy reads will get filtered out based on BLAST results and subsequent taxonomic assignment, since we’ll only be using Arthropoda and Alveolata reads.
SAMPLE 73
SAMPLE 113
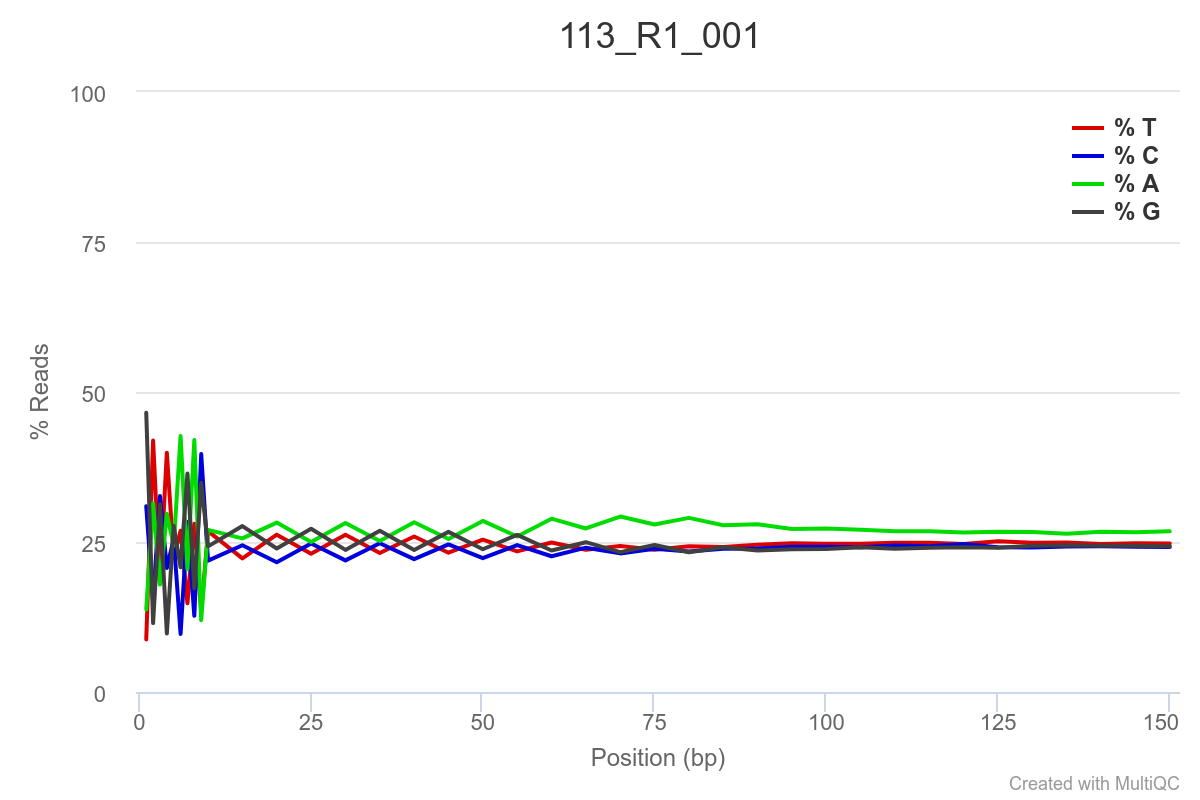

SAMPLE 118

SAMPLE 127
SAMPLE 222
SAMPLE 272


SAMPLE 280
SAMPLE 425

SAMPLE 427
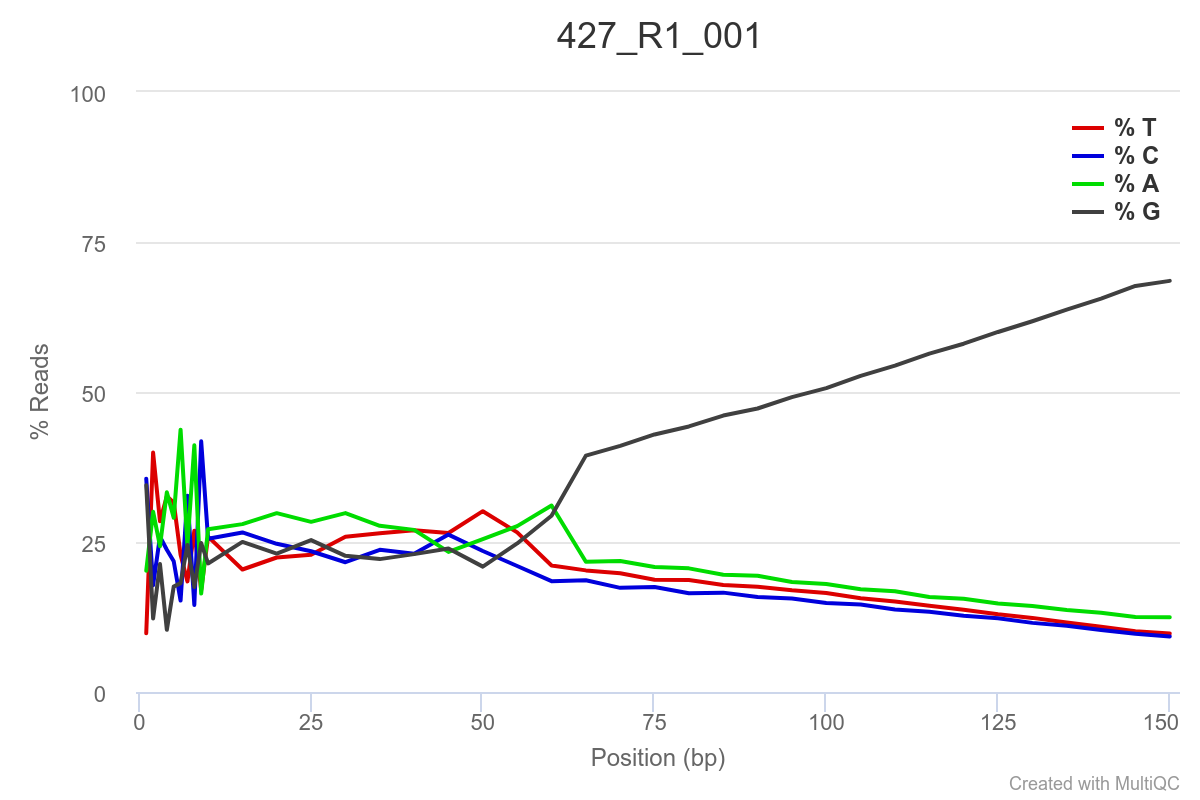
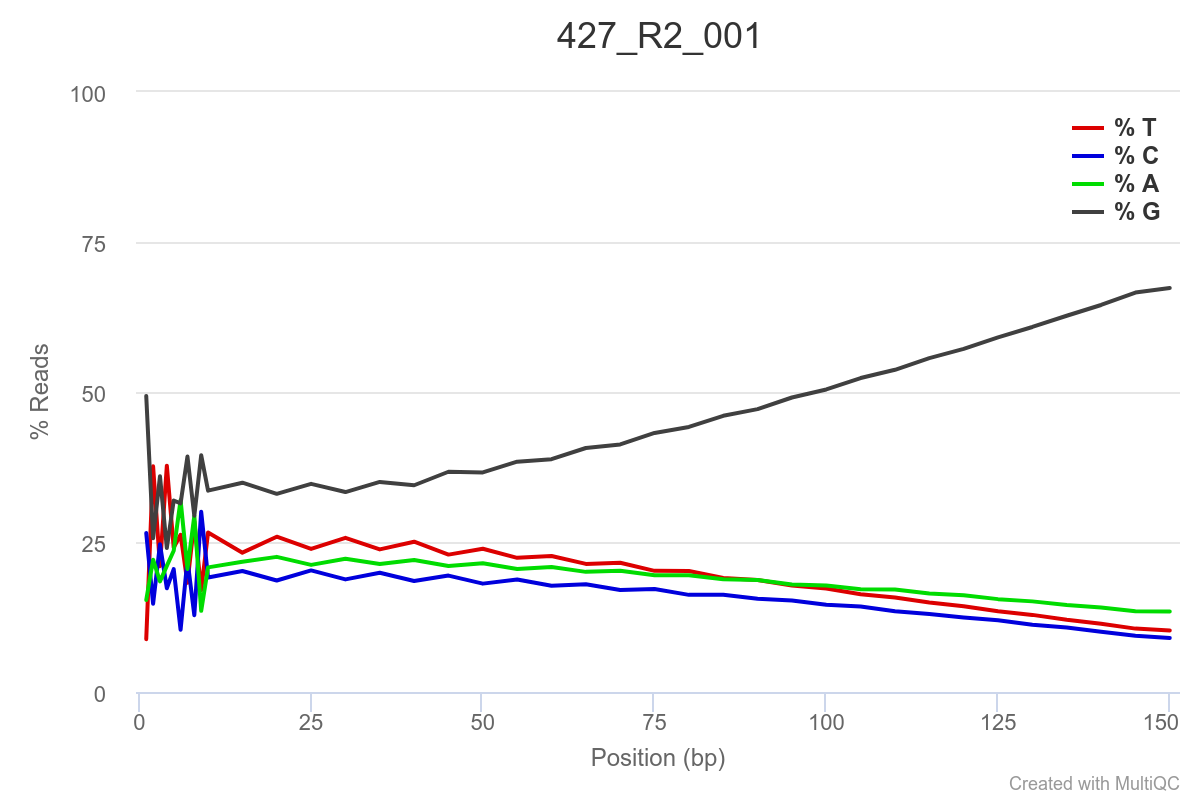
SAMPLE 445

SAMPLE 463
SAMPLE 481