Attempting to get some other metrics regarding our various C.bairdi transcriptome assemblies other than BUSCO scores, I decided to try running DETONATE, as this is a recommended tool by Trinity.
I recently ran DETONATE’s ref-eval (see 20200529) because it was relatively easy to do and thought it might be useful, as it’s included in the DETONATE package. However, the results are complicated to interpret and I’m not sure they actually tell us anything.
Continuing with the DETONATE package, I ran the other component (which is probably what we really want and will provide us with a simple “score” which will be easier to understand), rsem-eval. Plus, it’s a very low effort process, so might as well give it a whirl.
The job was run on Mox.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=cbai_detonate_transcriptome_evaluations
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=15-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20200616_cbai_detonate_transcriptome_evaluations
###################################################################################
# These variables need to be set by user
# Assign Variables
## frag_size is guesstimate of library fragment sizes
frag_size=500
reads_dir=/gscratch/srlab/sam/data/C_bairdi/RNAseq
transcriptomes_dir=/gscratch/srlab/sam/data/C_bairdi/transcriptomes
threads=28
# Array of the various comparisons to evaluate
# Each condition in each comparison should be separated by a "-"
transcriptomes_array=(
"${transcriptomes_dir}"/cbai_transcriptome_v1.0.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v1.5.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v1.6.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v1.7.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v2.0.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v2.1.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v3.0.fasta \
"${transcriptomes_dir}"/cbai_transcriptome_v3.1.fasta
)
###################################################################################
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Programs array
declare -A programs_array
programs_array=(
[bowtie2]="/gscratch/srlab/programs/bowtie2-2.3.5.1-linux-x86_64" \
[detonate_trans_length]="/gscratch/srlab/programs/detonate-1.11/rsem-eval/rsem-eval-estimate-transcript-length-distribution" \
[detonate]="/gscratch/srlab/programs/detonate-1.11/rsem-eval/rsem-eval-calculate-score"
)
# Loop through each comparison
for transcriptome in "${!transcriptomes_array[@]}"
do
## Inititalize arrays
R1_array=()
R2_array=()
reads_array=()
# Variables
R1_list=""
R2_list=""
transcriptome_name="${transcriptomes_array[$transcriptome]##*/}"
rsem_eval_dist_mean_sd="${transcriptome_name}_true_length_dis_mean_sd.txt"
# Capture FastA checksums for verification
echo "Generating checksum for ${transcriptome_name}"
md5sum "${transcriptomes_array[transcriptome]}" >> fasta.checksums.md5
echo "Finished generating checksum for ${transcriptome_name}"
echo ""
if [[ "${transcriptome_name}" == "cbai_transcriptome_v1.0.fasta" ]]; then
reads_array=("${reads_dir}"/20200[15][13][138]*megan*.fq)
# Create array of fastq R1 files
R1_array=("${reads_dir}"/20200[15][13][138]*megan*R1.fq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/20200[15][13][138]*megan*R2.fq)
elif [[ "${transcriptome_name}" == "cbai_transcriptome_v1.5.fasta" ]]; then
reads_array=("${reads_dir}"/20200[145][13][138]*megan*.fq)
# Create array of fastq R1 files
R1_array=("${reads_dir}"/20200[145][13][138]*megan*R1.fq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/20200[145][13][138]*megan*R2.fq)
elif [[ "${transcriptome_name}" == "cbai_transcriptome_v1.6.fasta" ]]; then
reads_array=("${reads_dir}"/*megan*.fq)
# Create array of fastq R1 files
R1_array=("${reads_dir}"/*megan*R1.fq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/*megan*R2.fq)
elif [[ "${transcriptome_name}" == "cbai_transcriptome_v1.7.fasta" ]]; then
reads_array=("${reads_dir}"/20200[145][13][189]*megan*.fq)
# Create array of fastq R1 files
R1_array=("${reads_dir}"/20200[145][13][189]*megan*R1.fq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/20200[145][13][189]*megan*R2.fq)
elif [[ "${transcriptome_name}" == "cbai_transcriptome_v2.0.fasta" ]] \
|| [[ "${transcriptome_name}" == "cbai_transcriptome_v2.1.fasta" ]]; then
reads_array=("${reads_dir}"/*fastp-trim*.fq)
# Create array of fastq R1 files
R1_array=("${reads_dir}"/*R1*fastp-trim*.fq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/*R2*fastp-trim*.fq)
elif [[ "${transcriptome_name}" == "cbai_transcriptome_v3.0.fasta" ]] \
|| [[ "${transcriptome_name}" == "cbai_transcriptome_v3.1.fasta" ]]; then
reads_array=("${reads_dir}"/*fastp-trim*20[12][09][01][24]1[48]*.fq)
# Create array of fastq R1 files
R1_array=("${reads_dir}"/*R1*fastp-trim*20[12][09][01][24]1[48]*.fq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/*R2*fastp-trim*20[12][09][01][24]1[48]*.fq)
fi
# Create list of fastq files used in analysis
## Uses parameter substitution to strip leading path from filename
printf "%s\n" "${reads_array[@]##*/}" >> "${transcriptome_name}".fastq.list.txt
# Create comma-separated lists of FastQ reads
R1_list=$(echo "${R1_array[@]}" | tr " " ",")
R2_list=$(echo "${R2_array[@]}" | tr " " ",")
# Determine transcript length
${programs_array[detonate_trans_length]} \
"${transcriptomes_array[$transcriptome]}" \
"${rsem_eval_dist_mean_sd}"
# Run rsem-eval
# Use bowtie2 and paired-end options
${programs_array[detonate]} \
--bowtie2 \
--bowtie2-path "${programs_array[bowtie2]}" \
--num-threads ${threads} \
--transcript-length-parameters "${rsem_eval_dist_mean_sd}" \
--paired-end \
"${R1_list}" \
"${R2_list}" \
"${transcriptomes_array[$transcriptome]}" \
"${transcriptome_name}" \
${frag_size}
done
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
# Capture program options
for program in "${!programs_array[@]}"
do
{
echo "Program options for ${program}: "
echo ""
${programs_array[$program]} --help
echo ""
echo ""
echo "----------------------------------------------"
echo ""
echo ""
} &>> program_options.log || true
done
RESULTS
This process was PAINFUL. Here’s the “runtime” for the failed job (due to it timing out; after 65 DAYS!). Also, this was just for cbai_transcriptome_v2.0!!!

Overall, even though this required very little effort on my part, it was kind of a pain to manage. For some reason (I guess it’s due to the number of sequences it has to align) cbai_transcriptome_v2.0 alignments took too long (i.e. longer than the 30 days between Mox node maintenance). I restarted this job a couple of times and I finally lucked out for a bit when the October, November, and December 2020 Mox maintenance dates were canceled. Despite that, I neglected to extend the runtime further in December and the job timed out.
But, with that said, part of me thinks something weird was going on anyway. I mean, look at the size of the BAM file (remember, a BAM file is a compressed version of a SAM file, and is usually close to 10x smaller than the originating SAM file!) that was still being made when the job died:
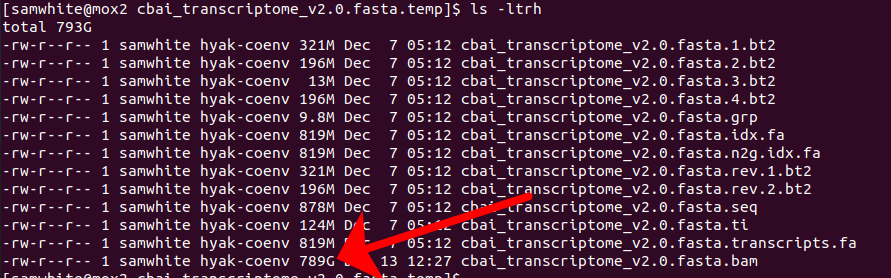
789GB!!!!
That’s absurd! Not to mention the fact that this was generated over the course of two months!
I’m going to try one more thing to see if I can get rsem-eval to work. Again, it’s low effort, so won’t take too much of my time. I’m going to run bowtie2 independently of rsem-eval (bowtie2 alignment is built-in to that, if the user wants to use it) and see if that is somehow faster. If it is, then I can provide the resulting BAM files as input to rsem-eval.