After noticing that the initial MEGAN6 taxonomic assignments for our combined C.bairdi NanoPore data from 20200917 revealed a high number of bases assigned to E.canceri and Aquifex sp., I decided to explore the taxonomic breakdown of just the individual samples to see which of the samples was contributing to these taxonomic assignments most.
Ran the Hematodinium-infected (6129_403_26_Q7) hemolymph NanoPore sequencing data through DIAMOND BLASTx (on Mox) and subsequent output conversion to the MEGAN6 RMA6 format (on swoose due to program Java X11 requirement which is not functional on Mox) to obtain taxonomic assignments.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=cbai_blastx_DIAMOND_nanopore_6129_403_26_Q7
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=10-00:00:00
## Memory per node
#SBATCH --mem=200G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20200928_cbai_diamond_blastx_nanopore_6129_403_26_Q7
# Script to run DIAMOND BLASTx on 6129_403_26 quality filtered (Q7) C.bairdi NanoPore reads
# from 20200928 using the --long-reads option
# for subsequent import into MEGAN6 to evaluate reads taxonomically.
###################################################################################
# These variables need to be set by user
# Input FastQ file
fastq=/gscratch/srlab/sam/data/C_bairdi/DNAseq/20200928_cbai_nanopore_6129_403_26_quality-7.fastq
# DIAMOND Output filename prefix
prefix=20200928_cbai_nanopore_6129_403_26_Q7
# Set number of CPUs to use
threads=28
# Program paths
diamond=/gscratch/srlab/programs/diamond-2.0.4/diamond
# DIAMOND NCBI nr database with taxonomy dumps
dmnd_db=/gscratch/srlab/blastdbs/ncbi-nr-20190925/nr.dmnd
###################################################################################
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# SegFault fix?
export THREADS_DAEMON_MODEL=1
# Inititalize arrays
programs_array=()
# Programs array
programs_array=("${diamond}")
md5sum "${fastq}" > fastq_checksums.md5
# Run DIAMOND with blastx
# Output format 6 produces a standard BLAST tab-delimited file
# Run DIAMOND with blastx
# Output format 100 produces a DAA binary file for use with MEGAN
${diamond} blastx \
--long-reads \
--db ${dmnd_db} \
--query "${fastq}" \
--out "${prefix}".blastx.daa \
--outfmt 100 \
--top 5 \
--block-size 8.0 \
--index-chunks 1 \
--threads ${threads}
# Capture program options
for program in "${!programs_array[@]}"
do
{
echo "Program options for ${programs_array[program]}: "
echo ""
${programs_array[program]} help
echo ""
echo ""
echo "----------------------------------------------"
echo ""
echo ""
} &>> program_options.log || true
done
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
Bash script (GitHub) for daa2rma on Emu:
#!/bin/bash
# Script to run MEGAN6 daa2rma on DIAMOND DAA files from
# 20200928_cbai_diamond_blastx_nanopore_6129_403_26_Q7.
# Utilizes the --longReads option
# Requires MEGAN mapping file from:
# http://ab.inf.uni-tuebingen.de/data/software/megan6/download/
# MEGAN mapping file
megan_map=/home/sam/data/databases/MEGAN6/megan-map-Jul2020-2.db
# Programs array
declare -A programs_array
programs_array=(
[daa2rma]="/home/shared/megan_6.19.9/tools/daa2rma"
)
threads=16
#########################################################################
# Exit script if any command fails
set -e
## Run daa2rma
# Capture start "time"
# Uses builtin bash variable called ${SECONDS}
start=${SECONDS}
for daa in *.daa
do
start_loop=${SECONDS}
sample_name=$(basename --suffix ".blastx.daa" "${daa}")
echo "Now processing ${sample_name}.daa2rma.rma6"
echo ""
# Run daa2rma with long reads option
${programs_array[daa2rma]} \
--in "${daa}" \
--longReads \
--mapDB ${megan_map} \
--out "${sample_name}".daa2rma.rma6 \
--threads ${threads} \
2>&1 | tee --append daa2rma_log.txt
end_loop=${SECONDS}
loop_runtime=$((end_loop-start_loop))
echo "Finished processing ${sample_name}.daa2rma.rma6 in ${loop_runtime} seconds."
echo ""
done
# Caputure end "time"
end=${SECONDS}
runtime=$((end-start))
# Print daa2rma runtime, in seconds
{
echo ""
echo "---------------------"
echo ""
echo "Total runtime was: ${runtime} seconds"
} >> daa2rma_log.txt
# Capture program options
for program in "${!programs_array[@]}"
do
{
echo "Program options for ${program}: "
echo ""
${programs_array[$program]} --help
echo ""
echo ""
echo "----------------------------------------------"
echo ""
echo ""
} &>> program_options.log || true
done
# Document programs in PATH
{
date
echo ""
echo "System PATH:"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
RESULTS
Pretty quick, a little over 31mins:

DIAMOND BLASTx Output folder:
Taxonomic tree
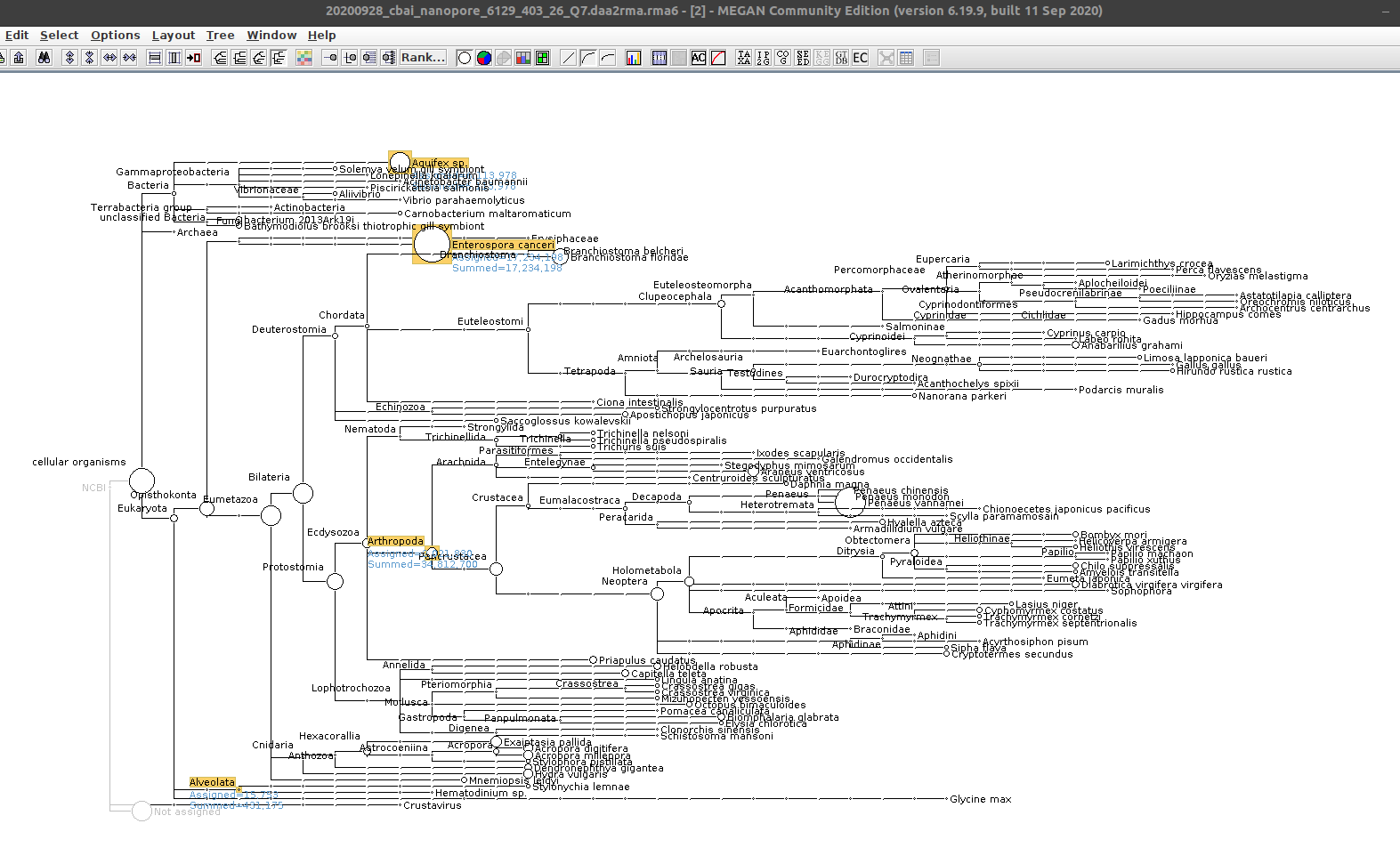
Alrighty, we see both Alveolata/Hematodinium sp., E.canceri_ Aquifex sp. taxonomic assignments (and, of course Arthropoda).
A large number of bases (~34Mbp) are assigned to Arthropoda. Alveolata only has ~400kbp assigned, while E.canceri has ~17Mbp (!) and Aquifex sp. has ~6Mbp!
Very surprising.