INTRO
This is the second day of a two day experiment. Day 0 corresponds to the first day of measurements, and Day 1 corresponds to the second day of measurements. The same clams were measured on both days. Day 0 notebook entry is available here:
Data and results are available here:
The section below was knitted from the R Markdown file 00.00-resazurin-20260401-clam-Day2-36C-C-vs-S.Rmd (GitHub).
1 Background
This is Day 2 of the experiment begun on 2026-03-31 (20260331-clam-36C-C-vs-S). After acute heat stress at 36°C on Day 1, resazurin solution was removed, plates were rinsed with sea water, and clams were stored overnight in the cold room. On Day 2 clams were again submerged in 5.0 mL resazurin working solution and returned to 36°C. Fluorescence was measured hourly (rather than every 30 min) on a Synergy HTX (Agilent) plate reader. This design allows assessment of metabolic recovery following repeated heat stress.
Plate layout was randomized; treatment assignments known only to Steven Roberts.
Important notes from this experiment:
- The following wells showed low volume and are flagged for exclusion in the layout: D A1, B A3, D B2, E C2.
- Plate A was slow to reach target temperature: wells ranged 11–12°C at T0, reaching 33–34°C only by T4.
See data/clam/20260401-clam-Day2-36C-C-vs-S/README.md for full experimental notes including per-timepoint temperature spot checks.
1.1 Expected inputs
| Path | Description |
|---|---|
data/clam/20260401-clam-Day2-36C-C-vs-S/plate-*-T*.txt |
Plate reader fluorescence exports (one file per plate per timepoint) |
data/clam/20260401-clam-Day2-36C-C-vs-S/layout.csv |
Well metadata: plate ID, well ID, blank flag, family/treatment groups, size measurements, exclusion flags |
1.2 Expected outputs
All outputs are written to output/clam/20260401-clam-Day2-36C-C-vs-S/.
| File | Description |
|---|---|
figures/ |
All plots generated by this script |
auc_all_metrics.csv |
Per-individual AUC values for every active measurement metric |
auc_summary.csv |
Group-level AUC summary statistics (mean, SD, SE, median) |
metabolism.csv |
Full per-well per-timepoint metabolism data frame |
pairwise_stats.csv |
Tukey-adjusted pairwise comparisons from AUC linear models |
2 Setup
knitr::opts_chunk$set(
echo = TRUE, # Display code chunks
eval = TRUE, # Evaluate code chunks
warning = FALSE, # Hide warnings
message = FALSE, # Hide messages
comment = "", # Prevents appending '##' to beginning of lines in code output
results = 'hold' # Holds output so it's all printed together after code chunk
)library(tidyverse)
library(pracma) # trapz()
library(lme4)
library(lmerTest)
library(emmeans)
library(multcompView)
library(cowplot)
library(colorspace) # qualitative_hcl() for large palettes3 Helper Functions
normalize_well_id <- function(x) {
x <- toupper(trimws(x))
valid <- str_detect(x, "^[A-Z]+[0-9]+$")
out <- rep(NA_character_, length(x))
if (!any(valid)) return(out)
m <- str_match(x[valid], "^([A-Z]+)([0-9]+)$")
out[valid] <- paste0(m[, 2], as.integer(m[, 3]))
out
}
parse_time_hr <- function(path) {
hit <- str_match(basename(path),
"(?i)-T([0-9]+(?:\\.[0-9]+)?)\\.txt$")
as.numeric(hit[, 2])
}
parse_plate_id <- function(path) {
hit <- str_match(basename(path),
"(?i)^plate-([A-Za-z0-9-]+)-T[0-9]+(?:\\.[0-9]+)?\\.txt$")
id <- hit[, 2]
ifelse(is.na(id), "unknown", id)
}
extract_results_block <- function(lines) {
results_idx <- which(trimws(lines) == "Results")
if (length(results_idx) == 0) stop("No Results section found")
idx <- results_idx[1]
header_tokens <- str_split(lines[idx + 1], "\\t")[[1]] |> trimws()
col_ids <- header_tokens[
header_tokens != "" & str_detect(header_tokens, "^[0-9]+$")]
j <- idx + 2
data_lines <- character()
while (j <= length(lines)) {
line <- lines[j]
if (trimws(line) == "") break
if (!str_detect(line, "^[A-Za-z]\\t")) break
data_lines <- c(data_lines, line)
j <- j + 1
}
list(col_ids = col_ids, data_lines = data_lines)
}
parse_plate_export <- function(path) {
lines <- readLines(path, warn = FALSE)
res <- extract_results_block(lines)
map_dfr(res$data_lines, function(line) {
tokens <- str_split(line, "\\t")[[1]] |> trimws()
tokens <- tokens[tokens != ""]
row_letter <- tokens[1]
nums <- suppressWarnings(as.numeric(tokens[-1]))
valid_idx <- which(!is.na(nums))
if (length(valid_idx) == 0) return(tibble())
vals <- nums[valid_idx]
n <- min(length(vals), length(res$col_ids))
tibble(
row_id = toupper(row_letter),
col_id = as.integer(res$col_ids[seq_len(n)]),
well_id = normalize_well_id(
paste0(toupper(row_letter), res$col_ids[seq_len(n)])),
value = vals[seq_len(n)]
)
}) %>%
mutate(
plate_id = str_to_lower(parse_plate_id(path)),
time_hr = parse_time_hr(path)
)
}
trapezoid_auc <- function(time_hr, value) {
ok <- is.finite(time_hr) & is.finite(value)
t <- time_hr[ok]
v <- value[ok]
if (length(t) < 2) return(NA_real_)
ord <- order(t)
t <- t[ord]; v <- v[ord]
sum(diff(t) * (head(v, -1) + tail(v, -1)) / 2)
}
# Shared helper: extract display unit string from a measurement column name.
# e.g. "area_mm2_measurement" -> "mm²", "weight_mg_measurement" -> "mg"
parse_meas_unit <- function(col_name) {
unit_raw <- col_name |>
str_remove("^metabolism_per_") |>
str_remove("_measurement$") |>
str_extract("[^_]+$")
case_when(
unit_raw == "mm2" ~ "mm²",
unit_raw == "cm2" ~ "cm²",
unit_raw == "mm3" ~ "mm³",
unit_raw == "cm3" ~ "cm³",
TRUE ~ unit_raw
)
}
# y-axis label for metabolism line plots: "fold change/mm²"
metabolism_y_label <- function(col_name) {
paste0("Metabolism (fold change/", parse_meas_unit(col_name), ")")
}
# y-axis label for AUC box plots: "Metabolism (AUC; mm²)"
auc_y_label <- function(metric_name) {
paste0("Metabolism (AUC; ", parse_meas_unit(metric_name), ")")
}4 Load Data
4.1 Plate export files
proj_root <- rprojroot::find_rstudio_root_file()
data_dir <- file.path(proj_root, "data", "clam",
"20260401-clam-Day2-36C-C-vs-S")
fig_dir <- file.path(proj_root, "output", "clam",
"20260401-clam-Day2-36C-C-vs-S", "figures")
out_dir <- file.path(proj_root, "output", "clam",
"20260401-clam-Day2-36C-C-vs-S")
dir.create(fig_dir, recursive = TRUE, showWarnings = FALSE)
dir.create(out_dir, recursive = TRUE, showWarnings = FALSE)
plate_files <- list.files(
data_dir,
pattern = "(?i)^plate-.*-T[0-9]+(?:\\.[0-9]+)?\\.txt$",
full.names = TRUE
)
plate_raw <- map_dfr(plate_files, function(path) {
tryCatch(parse_plate_export(path),
error = function(e) {
message("Parse error in ", basename(path), ": ", e$message)
tibble()
})
})
str(plate_raw)tibble [360 × 6] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:360] "A" "A" "A" "A" ...
$ col_id : int [1:360] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:360] "A1" "A2" "A3" "A4" ...
$ value : num [1:360] 583 360 898 455 421 ...
$ plate_id: chr [1:360] "a" "a" "a" "a" ...
$ time_hr : num [1:360] 0 0 0 0 0 0 0 0 0 0 ...4.2 Plate consistency check
Checks that every plate has the same number of wells at every timepoint. The expected well count is the mode across all plate × timepoint reads. Any plate with at least one deviating read is flagged and dropped entirely before any further analysis — removing only the aberrant timepoint would break the fold-change baseline calculation.
well_counts <- plate_raw %>%
group_by(plate_id, time_hr) %>%
summarise(n_wells = n_distinct(well_id), .groups = "drop")
expected_n_wells <- as.integer(
names(which.max(table(well_counts$n_wells)))
)
inconsistent_reads <- well_counts %>%
filter(n_wells != expected_n_wells) %>%
arrange(plate_id, time_hr)
inconsistent_plate_ids <- unique(inconsistent_reads$plate_id)
if (nrow(inconsistent_reads) > 0) {
cat("**Plate consistency check FAILED.**",
"Expected", expected_n_wells, "wells per plate-timepoint read.",
length(inconsistent_plate_ids),
"plate(s) have at least one deviating read and are excluded",
"from all analyses:\n\n")
cat(knitr::kable(
inconsistent_reads,
col.names = c("Plate", "Time (h)", "Wells read"),
caption = paste("Expected:", expected_n_wells, "wells per read")
), sep = "\n")
cat("\n")
plate_raw <- plate_raw %>%
filter(!plate_id %in% inconsistent_plate_ids)
message(length(inconsistent_plate_ids),
" plate(s) removed from plate_raw: ",
paste(inconsistent_plate_ids, collapse = ", "))
} else {
cat("Plate consistency check passed: all",
n_distinct(well_counts$plate_id), "plates have",
expected_n_wells, "wells at every timepoint.\n")
}Plate consistency check passed: all 6 plates have 12 wells at every timepoint.
4.3 Layout file
layout_path <- file.path(data_dir, "layout.csv")
layout_raw <- read_csv(layout_path,
col_types = cols(.default = "c"),
show_col_types = FALSE)
# Standardise column names to snake_case
names(layout_raw) <- names(layout_raw) |>
str_to_lower() |>
str_replace_all("[^a-z0-9]+", "_") |>
str_replace_all("_+", "_") |>
str_replace("_$", "")
# Normalise plate_id to match plate file ids (strip "plate-" prefix)
layout_clean <- layout_raw %>%
mutate(
plate_id = str_remove(str_to_lower(plate_id), "^plate-"),
well_id = normalize_well_id(plate_well),
is_blank = if ("is_blank" %in% names(layout_raw))
toupper(trimws(is_blank)) %in% c("TRUE", "T", "1", "YES", "Y")
else
FALSE
)
found_exclude_col <- intersect(
c("exclude_from_analysis", "exclude", "omit", "not_analyzed"),
names(layout_clean)
)[1]
layout_clean <- layout_clean %>%
mutate(
exclude_from_analysis = if (!is.na(found_exclude_col))
toupper(trimws(.data[[found_exclude_col]])) %in%
c("TRUE", "T", "1", "YES", "Y")
else
FALSE
)
# Identify measurement columns and group columns
measurement_cols <- names(layout_clean)[
str_detect(names(layout_clean), "_measurement$")]
group_cols <- names(layout_clean)[
str_detect(names(layout_clean), "_group$")]
# Cast measurement columns to numeric
layout_clean <- layout_clean %>%
mutate(across(all_of(measurement_cols),
~ suppressWarnings(as.numeric(.x))))
# Determine which measurement columns actually contain finite data
active_meas_cols <- measurement_cols[
sapply(measurement_cols, function(col)
any(is.finite(layout_clean[[col]]), na.rm = TRUE))]
# Normalise group values to lowercase so they match colour scale definitions
layout_clean <- layout_clean %>%
mutate(across(all_of(group_cols),
~ str_to_lower(trimws(as.character(.x)))))
message("Group columns: ", paste(group_cols, collapse = ", "))
message("Active measurement columns: ",
paste(active_meas_cols, collapse = ", "))
str(layout_clean)tibble [72 × 13] (S3: tbl_df/tbl/data.frame)
$ plate_id : chr [1:72] "a" "a" "a" "a" ...
$ plate_well : chr [1:72] "A01" "A02" "A03" "A04" ...
$ is_blank : logi [1:72] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:72] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:72] "1" "2" "3" "4" ...
$ treatment_group : chr [1:72] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:72] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:72] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:72] NA NA NA NA NA NA NA NA NA NA ...
$ area_mm2_measurement : num [1:72] 142.9 71.1 92.3 75.3 101.8 ...
$ imagej_id : chr [1:72] "1" "4" "3" "2" ...
$ well_id : chr [1:72] "A1" "A2" "A3" "A4" ...
$ exclude_from_analysis: logi [1:72] FALSE FALSE FALSE FALSE FALSE FALSE ...5 Merge Plate Data with Layout
dat <- plate_raw %>%
left_join(
layout_clean %>%
select(plate_id, well_id, is_blank, exclude_from_analysis,
any_of("exclude_reason"),
all_of(group_cols), all_of(measurement_cols)),
by = c("plate_id", "well_id")
) %>%
mutate(
is_blank = replace_na(is_blank, FALSE),
exclude_from_analysis = replace_na(exclude_from_analysis, FALSE)
)
str(dat)tibble [360 × 15] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:360] "A" "A" "A" "A" ...
$ col_id : int [1:360] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:360] "A1" "A2" "A3" "A4" ...
$ value : num [1:360] 583 360 898 455 421 ...
$ plate_id : chr [1:360] "a" "a" "a" "a" ...
$ time_hr : num [1:360] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:360] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:360] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:360] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:360] "1" "2" "3" "4" ...
$ treatment_group : chr [1:360] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:360] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:360] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:360] NA NA NA NA NA NA NA NA NA NA ...
$ area_mm2_measurement : num [1:360] 142.9 71.1 92.3 75.3 101.8 ...6 Raw Fluorescence
6.1 Data frame
# Wells in the plate reader output that have no layout entry get all-NA group
# columns after the join. Keep only wells assigned to at least one group.
active_gc <- intersect(group_cols, names(dat))
raw_df <- dat %>%
filter(
!is_blank,
if (length(active_gc) > 0)
if_any(all_of(active_gc), ~ !is.na(.))
else
TRUE
) %>%
mutate(
trace_id = if_else(
!is.na(sample_id_group) & trimws(as.character(sample_id_group)) != "",
as.character(sample_id_group),
paste(plate_id, well_id, sep = "_")
)
)
families <- sort(unique(na.omit(raw_df$family_id_group)))
treatments <- sort(unique(na.omit(raw_df$treatment_group)))
n_fam <- length(families)
n_trt <- length(treatments)
# Palette strategy:
# <= 7 groups : Okabe-Ito (gold standard for colorblind-safe figures).
# > 7 groups : colorspace::qualitative_hcl("Dynamic") scales to any N
# using perceptually uniform HCL space — no colour collisions.
# Black (#000000) is excluded from both and reserved for blank wells.
okabe_ito_7 <- c(
"#E69F00", "#56B4E9", "#009E73", "#F0E442",
"#0072B2", "#D55E00", "#CC79A7"
)
make_palette <- function(n) {
if (n == 0L) return(character(0))
if (n <= length(okabe_ito_7)) return(okabe_ito_7[seq_len(n)])
colorspace::qualitative_hcl(n, palette = "Dynamic")
}
all_colours <- make_palette(n_fam + n_trt)
fam_colours <- setNames(all_colours[seq_len(n_fam)], families)
trt_colours <- setNames(all_colours[n_fam + seq_len(n_trt)], treatments)
lty_pool <- c("solid", "dashed", "dotted", "dotdash", "longdash")
trt_linetypes <- setNames(
lty_pool[(seq_len(n_trt) - 1L) %% length(lty_pool) + 1L],
treatments
)
plate_well_colours <- c(blank = "black", fam_colours)
str(raw_df)tibble [330 × 16] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:330] "A" "A" "A" "A" ...
$ col_id : int [1:330] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:330] "A1" "A2" "A3" "A4" ...
$ value : num [1:330] 583 360 898 455 421 ...
$ plate_id : chr [1:330] "a" "a" "a" "a" ...
$ time_hr : num [1:330] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:330] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:330] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:330] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:330] "1" "2" "3" "4" ...
$ treatment_group : chr [1:330] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ area_mm2_measurement : num [1:330] 142.9 71.1 92.3 75.3 101.8 ...
$ trace_id : chr [1:330] "1" "2" "3" "4" ...6.2 Raw fluorescence by plate (including blanks)
p_raw_plates <- dat %>%
filter(is.finite(time_hr), is.finite(value)) %>%
mutate(
colour_group = if_else(is_blank, "blank",
coalesce(family_id_group, "sample")),
trace_id = paste(plate_id, well_id, sep = "_")
) %>%
ggplot(aes(x = time_hr, y = value,
group = trace_id, colour = colour_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1, alpha = 0.7) +
facet_wrap(~ plate_id) +
scale_colour_manual(
values = plate_well_colours,
name = "Group",
breaks = names(plate_well_colours),
na.value = "grey80"
) +
labs(x = "Time (h)", y = "Raw fluorescence (RFU)") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_raw_plates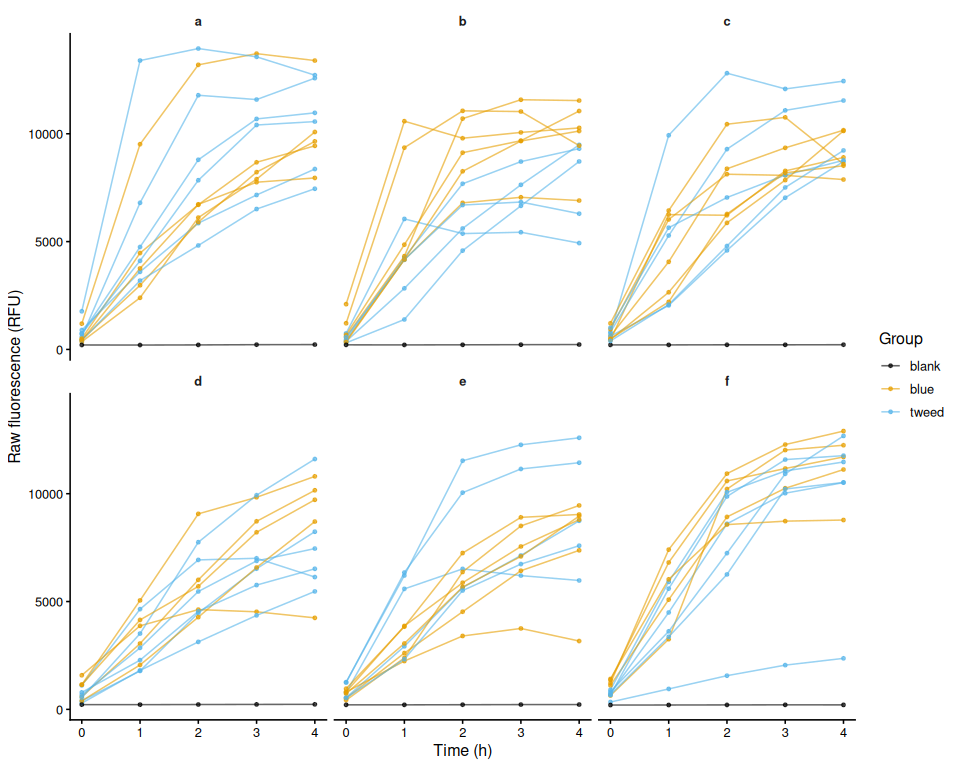
ggsave(file.path(fig_dir, "raw_fluor_by_plate.png"),
p_raw_plates, width = 10, height = 8)6.3 Mean raw fluorescence by family
raw_family_summary <- raw_df %>%
group_by(family_id_group, treatment_group, time_hr) %>%
summarise(
mean_fluor = mean(value, na.rm = TRUE),
se_fluor = sd(value, na.rm = TRUE) /
sqrt(sum(!is.na(value))),
n = sum(!is.na(value)),
.groups = "drop"
)
p_raw_mean <- ggplot(raw_family_summary,
aes(x = time_hr, y = mean_fluor,
colour = family_id_group, linetype = treatment_group,
group = interaction(family_id_group, treatment_group))) +
geom_ribbon(aes(ymin = mean_fluor - se_fluor,
ymax = mean_fluor + se_fluor,
fill = family_id_group),
alpha = 0.15, colour = NA) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_colour_manual(values = fam_colours, name = "Family") +
scale_fill_manual(values = fam_colours, name = "Family") +
scale_linetype_manual(values = trt_linetypes, name = "Treatment") +
labs(x = "Time (h)", y = "Mean raw fluorescence (RFU ± SE)") +
theme_classic(base_size = 13)
p_raw_mean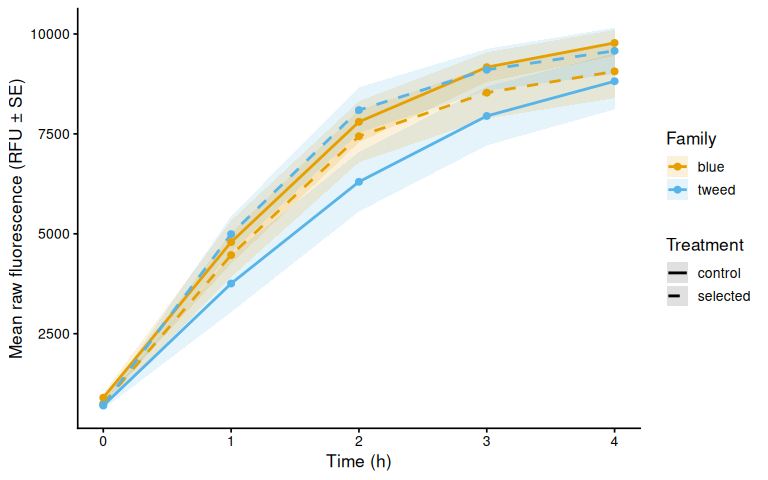
ggsave(file.path(fig_dir, "raw_mean_by_family.png"),
p_raw_mean, width = 8, height = 5)6.4 Individual raw fluorescence traces by family
p_raw_by_family <- raw_df %>%
ggplot(aes(x = time_hr, y = value,
group = trace_id, colour = treatment_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ family_id_group) +
scale_colour_manual(values = trt_colours, name = "Treatment") +
labs(x = "Time (h)", y = "Raw fluorescence (RFU)") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_raw_by_family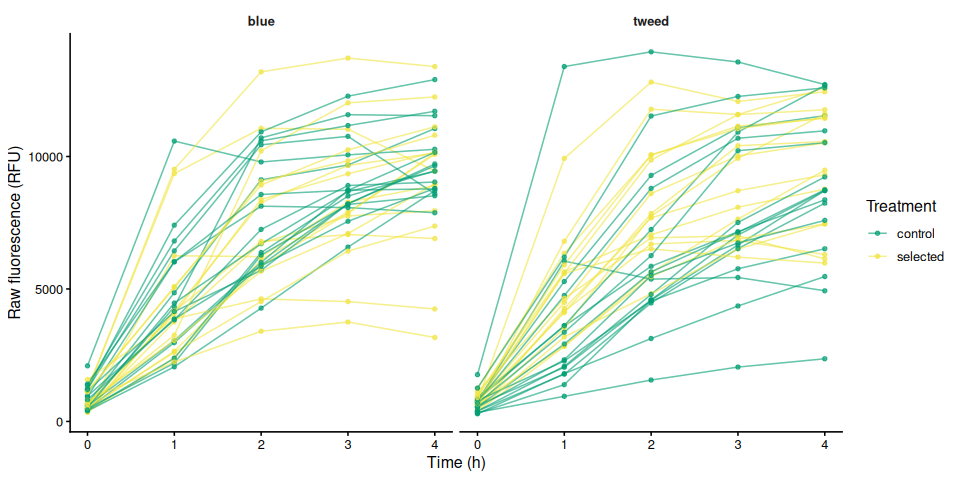
ggsave(file.path(fig_dir, "raw_individual_by_family.png"),
p_raw_by_family, width = 10, height = 5)6.5 Individual raw fluorescence traces by treatment
p_raw_by_treatment <- raw_df %>%
ggplot(aes(x = time_hr, y = value,
group = trace_id, colour = family_id_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ treatment_group) +
scale_colour_manual(values = fam_colours, name = "Family") +
labs(x = "Time (h)", y = "Raw fluorescence (RFU)") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_raw_by_treatment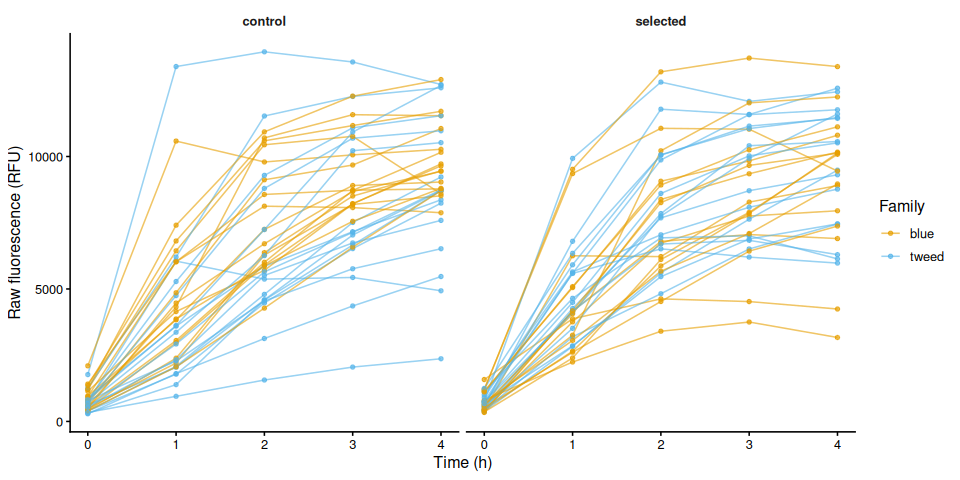
ggsave(file.path(fig_dir, "raw_individual_by_treatment.png"),
p_raw_by_treatment, width = 10, height = 5)6.6 Excluded samples
Wells flagged exclude_from_analysis = TRUE appear in the raw fluorescence plots above but are omitted from all analyses that follow.
excluded_wells <- dat %>%
filter(!is_blank, exclude_from_analysis) %>%
mutate(
sample = if_else(
!is.na(sample_id_group) & trimws(as.character(sample_id_group)) != "",
as.character(sample_id_group),
paste(plate_id, well_id, sep = "_")
)
) %>%
select(plate_id, well_id, sample, family_id_group, treatment_group,
any_of("exclude_reason")) %>%
distinct() %>%
arrange(plate_id, well_id)
if (nrow(excluded_wells) > 0) {
col_names <- c("Plate", "Well", "Sample", "Family", "Treatment")
if ("exclude_reason" %in% names(excluded_wells))
col_names <- c(col_names, "Reason")
cat(knitr::kable(excluded_wells, col.names = col_names), sep = "\n")
} else {
cat("No wells are excluded from analysis.\n")
}No wells are excluded from analysis.
7 Blank Correction via Fold-Change Normalization
Following Huffmyer et al.: fluorescence is first expressed as fold-change relative to each well’s own T0 reading (applied to samples and blanks alike), the mean fold-change of blank wells (per plate, per timepoint) is then subtracted. All samples therefore start at exactly 0 at T0 by construction, eliminating the risk of negative starting values from pipetting variance.
7.1 Step 1 – Fold-change relative to T0 for all wells
t0_all <- dat %>%
filter(is.finite(time_hr), is.finite(value)) %>%
group_by(plate_id, well_id) %>%
slice_min(time_hr, n = 1, with_ties = FALSE) %>%
select(plate_id, well_id, value_t0 = value) %>%
ungroup()
dat_fc <- dat %>%
left_join(t0_all, by = c("plate_id", "well_id")) %>%
mutate(fold_change = if_else(
is.finite(value_t0) & value_t0 > 0,
value / value_t0,
NA_real_
))
str(dat_fc)tibble [360 × 17] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:360] "A" "A" "A" "A" ...
$ col_id : int [1:360] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:360] "A1" "A2" "A3" "A4" ...
$ value : num [1:360] 583 360 898 455 421 ...
$ plate_id : chr [1:360] "a" "a" "a" "a" ...
$ time_hr : num [1:360] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:360] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:360] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:360] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:360] "1" "2" "3" "4" ...
$ treatment_group : chr [1:360] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:360] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:360] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:360] NA NA NA NA NA NA NA NA NA NA ...
$ area_mm2_measurement : num [1:360] 142.9 71.1 92.3 75.3 101.8 ...
$ value_t0 : num [1:360] 583 360 898 455 421 ...
$ fold_change : num [1:360] 1 1 1 1 1 1 1 1 1 1 ...7.2 Step 2 – Mean blank fold-change per plate per timepoint
blank_fc_ref <- dat_fc %>%
filter(is_blank) %>%
group_by(plate_id, time_hr) %>%
summarise(mean_blank_fc = mean(fold_change, na.rm = TRUE),
.groups = "drop")
str(blank_fc_ref)tibble [30 × 3] (S3: tbl_df/tbl/data.frame)
$ plate_id : chr [1:30] "a" "a" "a" "a" ...
$ time_hr : num [1:30] 0 1 2 3 4 0 1 2 3 4 ...
$ mean_blank_fc: num [1:30] 1 0.986 1.014 1.068 1.087 ...7.3 Step 3 – Subtract blank fold-change from sample fold-change
samples <- dat_fc %>%
filter(!is_blank, !exclude_from_analysis) %>%
mutate(
trace_id = if_else(
!is.na(sample_id_group) & trimws(as.character(sample_id_group)) != "",
as.character(sample_id_group),
paste(plate_id, well_id, sep = "_")
)
) %>%
left_join(blank_fc_ref, by = c("plate_id", "time_hr")) %>%
mutate(corrected_fc = fold_change - mean_blank_fc)
str(samples)tibble [330 × 20] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:330] "A" "A" "A" "A" ...
$ col_id : int [1:330] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:330] "A1" "A2" "A3" "A4" ...
$ value : num [1:330] 583 360 898 455 421 ...
$ plate_id : chr [1:330] "a" "a" "a" "a" ...
$ time_hr : num [1:330] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:330] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:330] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:330] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:330] "1" "2" "3" "4" ...
$ treatment_group : chr [1:330] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ area_mm2_measurement : num [1:330] 142.9 71.1 92.3 75.3 101.8 ...
$ value_t0 : num [1:330] 583 360 898 455 421 ...
$ fold_change : num [1:330] 1 1 1 1 1 1 1 1 1 1 ...
$ trace_id : chr [1:330] "1" "2" "3" "4" ...
$ mean_blank_fc : num [1:330] 1 1 1 1 1 1 1 1 1 1 ...
$ corrected_fc : num [1:330] 0 0 0 0 0 0 0 0 0 0 ...8 Blank-Corrected Fold-Change
8.1 Mean by family
bc_fc_summary <- samples %>%
group_by(family_id_group, treatment_group, time_hr) %>%
summarise(
mean_val = mean(corrected_fc, na.rm = TRUE),
se_val = sd(corrected_fc, na.rm = TRUE) /
sqrt(sum(!is.na(corrected_fc))),
n = sum(!is.na(corrected_fc)),
.groups = "drop"
)
p_bc_fc_mean <- ggplot(bc_fc_summary,
aes(x = time_hr, y = mean_val,
colour = family_id_group, linetype = treatment_group,
group = interaction(family_id_group, treatment_group))) +
geom_ribbon(aes(ymin = mean_val - se_val,
ymax = mean_val + se_val,
fill = family_id_group),
alpha = 0.15, colour = NA) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_colour_manual(values = fam_colours, name = "Family") +
scale_fill_manual(values = fam_colours, name = "Family") +
scale_linetype_manual(values = trt_linetypes, name = "Treatment") +
labs(x = "Time (h)",
y = "Mean blank-corrected fold-change (± SE)") +
theme_classic(base_size = 13)
p_bc_fc_mean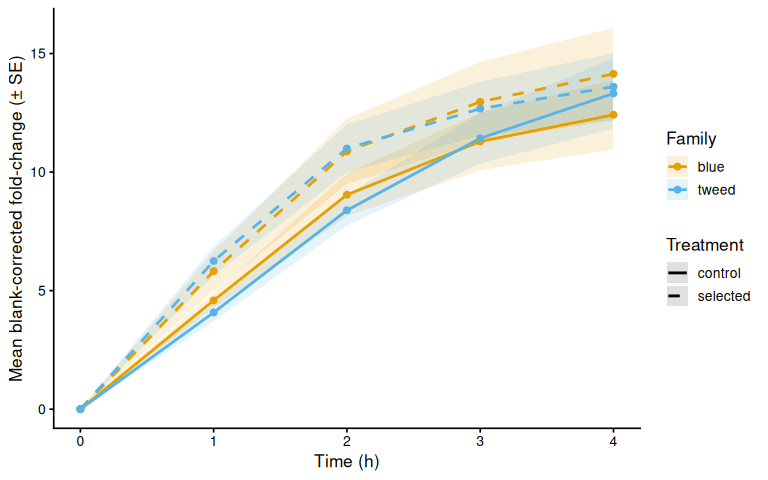
ggsave(file.path(fig_dir, "blank_corrected_fc_mean_by_family.png"),
p_bc_fc_mean, width = 8, height = 5)8.2 Individual traces by family
p_bc_fc_by_family <- samples %>%
ggplot(aes(x = time_hr, y = corrected_fc,
group = trace_id, colour = treatment_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ family_id_group) +
scale_colour_manual(values = trt_colours, name = "Treatment") +
labs(x = "Time (h)", y = "Blank-corrected fold-change") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_bc_fc_by_family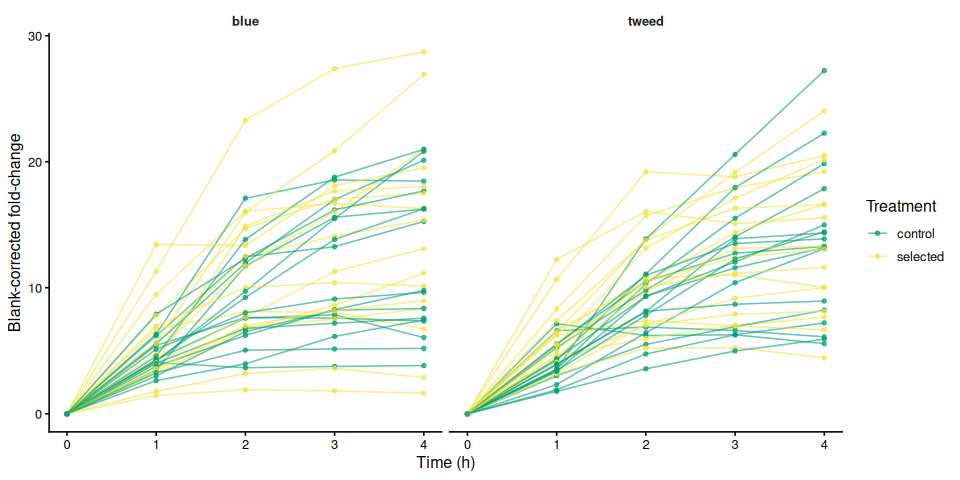
ggsave(file.path(fig_dir, "blank_corrected_fc_by_family.png"),
p_bc_fc_by_family, width = 10, height = 5)8.3 Individual blank-corrected fold-change traces by treatment
p_bc_fc_by_treatment <- samples %>%
ggplot(aes(x = time_hr, y = corrected_fc,
group = trace_id, colour = family_id_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ treatment_group) +
scale_colour_manual(values = fam_colours, name = "Family") +
labs(x = "Time (h)", y = "Blank-corrected fold-change") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_bc_fc_by_treatment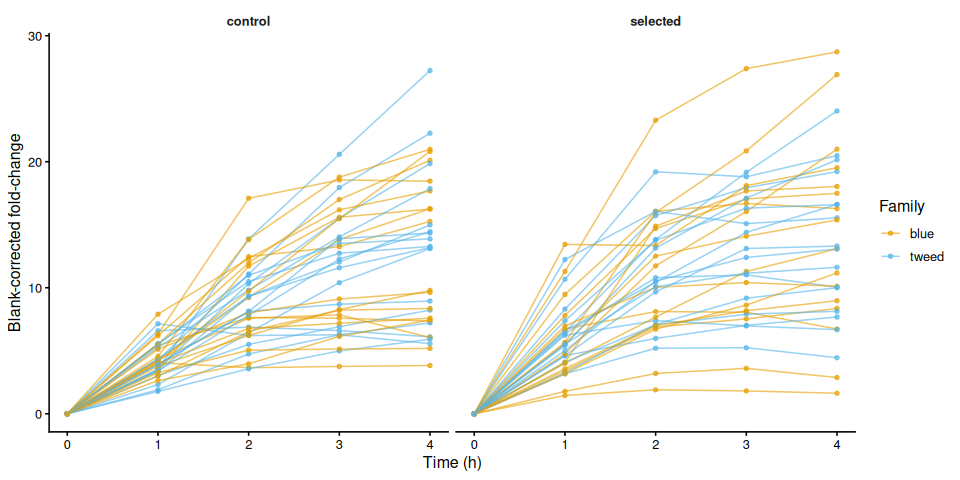
ggsave(file.path(fig_dir, "blank_corrected_fc_by_treatment.png"),
p_bc_fc_by_treatment, width = 10, height = 5)9 Metabolism (Size-Normalised Fold-Change)
Blank-corrected fold-change divided by each active measurement column. This is “metabolism” as defined in Huffmyer et al.
if (length(active_meas_cols) == 0) {
message("No active measurement columns: skipping metabolism calculation.")
metabolism_df <- tibble()
} else {
metabolism_df <- samples
for (mc in active_meas_cols) {
out_col <- paste0("metabolism_per_", mc)
metabolism_df <- metabolism_df %>%
mutate(!!out_col := if_else(
is.finite(.data[[mc]]) & .data[[mc]] > 0 &
is.finite(corrected_fc),
corrected_fc / .data[[mc]],
NA_real_
))
}
}
str(metabolism_df)tibble [330 × 21] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:330] "A" "A" "A" "A" ...
$ col_id : int [1:330] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:330] "A1" "A2" "A3" "A4" ...
$ value : num [1:330] 583 360 898 455 421 ...
$ plate_id : chr [1:330] "a" "a" "a" "a" ...
$ time_hr : num [1:330] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:330] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis : logi [1:330] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:330] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:330] "1" "2" "3" "4" ...
$ treatment_group : chr [1:330] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement : num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement : num [1:330] NA NA NA NA NA NA NA NA NA NA ...
$ area_mm2_measurement : num [1:330] 142.9 71.1 92.3 75.3 101.8 ...
$ value_t0 : num [1:330] 583 360 898 455 421 ...
$ fold_change : num [1:330] 1 1 1 1 1 1 1 1 1 1 ...
$ trace_id : chr [1:330] "1" "2" "3" "4" ...
$ mean_blank_fc : num [1:330] 1 1 1 1 1 1 1 1 1 1 ...
$ corrected_fc : num [1:330] 0 0 0 0 0 0 0 0 0 0 ...
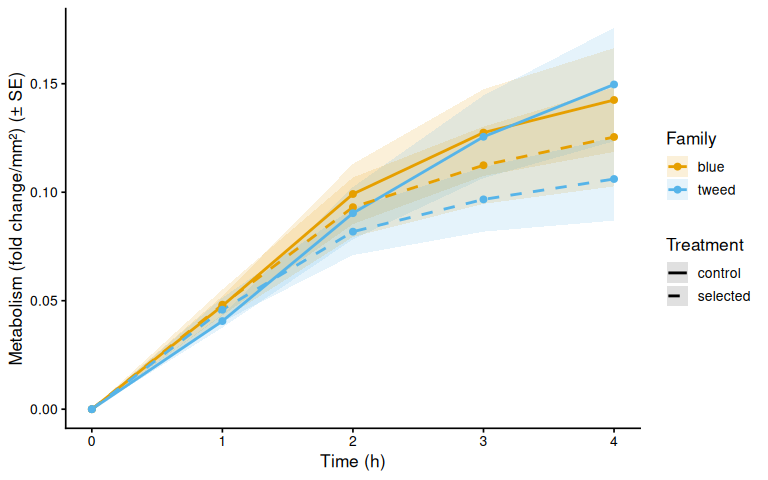
$ metabolism_per_area_mm2_measurement: num [1:330] 0 0 0 0 0 0 0 0 0 0 ...9.1 Mean metabolism by family
if (nrow(metabolism_df) > 0) {
metab_cols <- paste0("metabolism_per_", active_meas_cols)
for (col in metab_cols) {
if (!col %in% names(metabolism_df)) next
mc_label <- str_remove(col, "^metabolism_per_")
metab_summary <- metabolism_df %>%
group_by(family_id_group, treatment_group, time_hr) %>%
summarise(
mean_val = mean(.data[[col]], na.rm = TRUE),
se_val = sd(.data[[col]], na.rm = TRUE) /
sqrt(sum(!is.na(.data[[col]]))),
.groups = "drop"
)
p_metab_mean <- ggplot(metab_summary,
aes(x = time_hr, y = mean_val,
colour = family_id_group, linetype = treatment_group,
group = interaction(family_id_group, treatment_group))) +
geom_ribbon(aes(ymin = mean_val - se_val,
ymax = mean_val + se_val,
fill = family_id_group),
alpha = 0.15, colour = NA) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_colour_manual(values = fam_colours, name = "Family") +
scale_fill_manual(values = fam_colours, name = "Family") +
scale_linetype_manual(values = trt_linetypes, name = "Treatment") +
labs(x = "Time (h)",
y = paste0(metabolism_y_label(col), " (± SE)")) +
theme_classic(base_size = 13)
print(p_metab_mean)
ggsave(
file.path(fig_dir,
paste0("metabolism_mean_", mc_label, ".png")),
p_metab_mean, width = 8, height = 5)
}
}
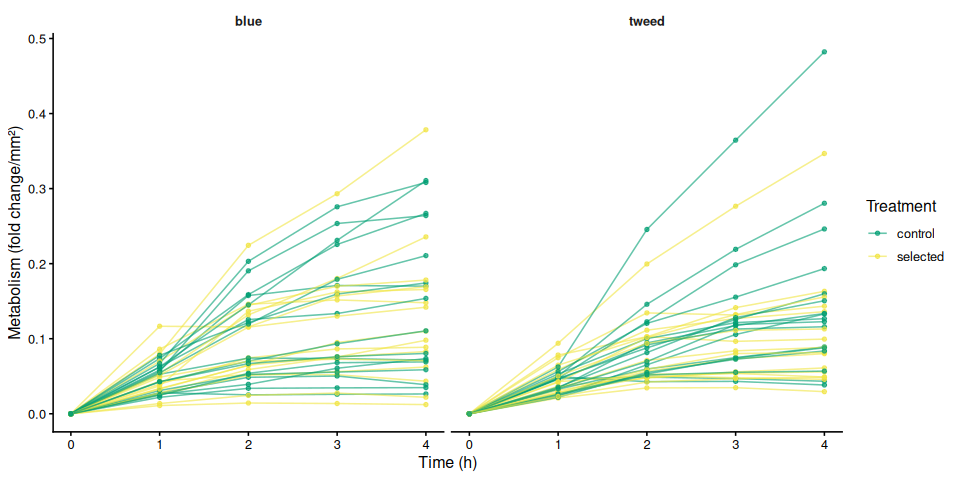
9.2 Individual metabolism traces by family
if (nrow(metabolism_df) > 0) {
for (col in metab_cols) {
if (!col %in% names(metabolism_df)) next
mc_label <- str_remove(col, "^metabolism_per_")
p_metab_by_family <- ggplot(metabolism_df,
aes(x = time_hr, y = .data[[col]],
group = trace_id, colour = treatment_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ family_id_group) +
scale_colour_manual(values = trt_colours, name = "Treatment") +
labs(x = "Time (h)", y = metabolism_y_label(col)) +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
print(p_metab_by_family)
ggsave(
file.path(fig_dir,
paste0("metabolism_individual_", mc_label, "_by_family.png")),
p_metab_by_family, width = 10, height = 5)
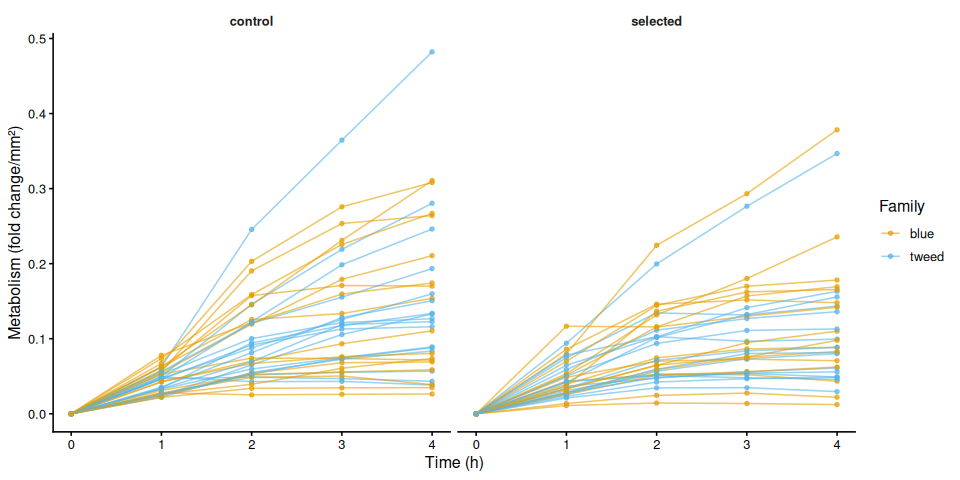
p_metab_by_treatment <- ggplot(metabolism_df,
aes(x = time_hr, y = .data[[col]],
group = trace_id, colour = family_id_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ treatment_group) +
scale_colour_manual(values = fam_colours, name = "Family") +
labs(x = "Time (h)", y = metabolism_y_label(col)) +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
print(p_metab_by_treatment)
ggsave(
file.path(fig_dir,
paste0("metabolism_individual_", mc_label, "_by_treatment.png")),
p_metab_by_treatment, width = 10, height = 5)
}
}
10 Time-Series Statistical Analysis
Linear mixed effects models test the effect of experimental variables on metabolism over time. Individual (sample_id_group) is included as a random intercept to account for repeated measures across timepoints. Type III ANOVA with Satterthwaite’s approximation (lmerTest) assesses significance; post-hoc pairwise comparisons use estimated marginal means (emmeans, Tukey adjustment).
run_ts_stats <- function(df, value_col) {
has_family <- "family_id_group" %in% names(df) &&
length(unique(na.omit(df$family_id_group))) > 1
has_treatment <- "treatment_group" %in% names(df) &&
length(unique(na.omit(df$treatment_group))) > 1
if (!has_family && !has_treatment) return(NULL)
df <- df %>%
filter(is.finite(.data[[value_col]]), is.finite(time_hr)) %>%
mutate(
time_f = factor(time_hr),
individual = factor(trace_id)
)
if (nrow(df) == 0) return(NULL)
if (has_family) df <- df %>% mutate(family = factor(family_id_group))
if (has_treatment) df <- df %>% mutate(treatment = factor(treatment_group))
if (has_family && length(unique(na.omit(df$family))) < 2) return(NULL)
if (has_treatment && length(unique(na.omit(df$treatment))) < 2) return(NULL)
fixed <- if (has_family && has_treatment)
paste0(value_col, " ~ time_f * family * treatment")
else if (has_family)
paste0(value_col, " ~ time_f * family")
else
paste0(value_col, " ~ time_f * treatment")
model <- lmer(
as.formula(paste0(fixed, " + (1 | individual)")),
data = df
)
anova_res <- anova(model, type = 3, ddf = "Satterthwaite")
# Pairwise comparisons of group combinations at each timepoint
emm_spec <- if (has_family && has_treatment)
~ family * treatment | time_f
else if (has_family)
~ family | time_f
else
~ treatment | time_f
emm <- emmeans(model, emm_spec)
pairs_res <- as.data.frame(pairs(emm, adjust = "tukey"))
# Main-effect marginal means (collapsed across time)
emm_main <- if (has_family && has_treatment)
emmeans(model, ~ family * treatment)
else if (has_family)
emmeans(model, ~ family)
else
emmeans(model, ~ treatment)
pairs_main <- as.data.frame(pairs(emm_main, adjust = "tukey"))
list(
model = model,
anova = anova_res,
pairs_by_time = pairs_res,
pairs_main = pairs_main,
has_family = has_family,
has_treatment = has_treatment
)
}
ts_stats <- list()
if (nrow(metabolism_df) > 0) {
for (mc in active_meas_cols) {
col <- paste0("metabolism_per_", mc)
if (col %in% names(metabolism_df))
ts_stats[[col]] <- run_ts_stats(metabolism_df, col)
}
}10.1 Results
for (col in names(ts_stats)) {
res <- ts_stats[[col]]
if (is.null(res)) next
cat("\n\n----\n### Metric:", col, "\n\n")
cat("**Type III ANOVA (Satterthwaite approximation):**\n")
print(res$anova)
cat("\n**Marginal means – main effects (collapsed across time):**\n")
print(res$pairs_main)
cat("\n**Pairwise comparisons by timepoint (Tukey):**\n")
print(res$pairs_by_time)
}Metric: metabolism_per_area_mm2_measurement
Signif. codes: 0 ‘’ 0.001 ’’ 0.01 ’’ 0.05 ‘.’ 0.1 ’ ’ 1
Marginal means – main effects (collapsed across time): contrast estimate SE df t.ratio p.value blue control - tweed control 0.002124428 0.01598627 62 0.133 0.9992 blue control - blue selected 0.007526561 0.01623413 62 0.464 0.9667 blue control - tweed selected 0.017269638 0.01623413 62 1.064 0.7126 tweed control - blue selected 0.005402134 0.01623413 62 0.333 0.9872 tweed control - tweed selected 0.015145211 0.01623413 62 0.933 0.7873 blue selected - tweed selected 0.009743077 0.01647826 62 0.591 0.9344
Results are averaged over the levels of: time_f Degrees-of-freedom method: kenward-roger P value adjustment: tukey method for comparing a family of 4 estimates
Pairwise comparisons by timepoint (Tukey): time_f = 0: contrast estimate SE df t.ratio p.value blue control - tweed control 0.00000000 0.02027045 146.72 0.000 1.0000 blue control - blue selected 0.00000000 0.02058473 146.72 0.000 1.0000 blue control - tweed selected 0.00000000 0.02058473 146.72 0.000 1.0000 tweed control - blue selected 0.00000000 0.02058473 146.72 0.000 1.0000 tweed control - tweed selected 0.00000000 0.02058473 146.72 0.000 1.0000 blue selected - tweed selected 0.00000000 0.02089430 146.72 0.000 1.0000
time_f = 1: contrast estimate SE df t.ratio p.value blue control - tweed control 0.00703033 0.02027045 146.72 0.347 0.9856 blue control - blue selected -0.00052600 0.02058473 146.72 -0.026 1.0000 blue control - tweed selected 0.00179666 0.02058473 146.72 0.087 0.9998 tweed control - blue selected -0.00755634 0.02058473 146.72 -0.367 0.9830 tweed control - tweed selected -0.00523367 0.02058473 146.72 -0.254 0.9942 blue selected - tweed selected 0.00232267 0.02089430 146.72 0.111 0.9995
time_f = 2: contrast estimate SE df t.ratio p.value blue control - tweed control 0.00883193 0.02027045 146.72 0.436 0.9722 blue control - blue selected 0.00607473 0.02058473 146.72 0.295 0.9910 blue control - tweed selected 0.01735596 0.02058473 146.72 0.843 0.8337 tweed control - blue selected -0.00275719 0.02058473 146.72 -0.134 0.9991 tweed control - tweed selected 0.00852404 0.02058473 146.72 0.414 0.9760 blue selected - tweed selected 0.01128123 0.02089430 146.72 0.540 0.9491
time_f = 3: contrast estimate SE df t.ratio p.value blue control - tweed control 0.00191613 0.02027045 146.72 0.095 0.9997 blue control - blue selected 0.01504467 0.02058473 146.72 0.731 0.8846 blue control - tweed selected 0.03078463 0.02058473 146.72 1.496 0.4428 tweed control - blue selected 0.01312855 0.02058473 146.72 0.638 0.9196 tweed control - tweed selected 0.02886850 0.02058473 146.72 1.402 0.4999 blue selected - tweed selected 0.01573996 0.02089430 146.72 0.753 0.8751
time_f = 4: contrast estimate SE df t.ratio p.value blue control - tweed control -0.00715625 0.02027045 146.72 -0.353 0.9849 blue control - blue selected 0.01703941 0.02058473 146.72 0.828 0.8412 blue control - tweed selected 0.03641093 0.02058473 146.72 1.769 0.2925 tweed control - blue selected 0.02419565 0.02058473 146.72 1.175 0.6433 tweed control - tweed selected 0.04356718 0.02058473 146.72 2.116 0.1527 blue selected - tweed selected 0.01937153 0.02089430 146.72 0.927 0.7904
Degrees-of-freedom method: kenward-roger P value adjustment: tukey method for comparing a family of 4 estimates
Area Under the Curve (AUC)
AUC computed per individual via the trapezoid rule across all timepoints. metabolism_per_* is the primary metric matching the paper; corrected_fc and raw_fluorescence are retained for reference.
compute_auc <- function(df, value_col, group_vars) { df %>% filter(is.finite(time_hr), is.finite(.data[[value_col]])) %>% group_by(across(all_of(group_vars))) %>% summarise( AUC = trapezoid_auc(time_hr, .data[[value_col]]), n_timepoints = n(), .groups = “drop” ) %>% filter(is.finite(AUC)) }
\# Only include grouping columns that are actually present in the data individual_vars <- intersect( c(“trace_id”, “family_id_group”, “treatment_group”), names(metabolism_df) )
auc_metab_list <- list() if (nrow(metabolism_df) > 0) { for (mc in active_meas_cols) { col <- paste0(“metabolism_per_”, mc) if (col %in% names(metabolism_df)) { auc_metab_list[[col]] <- compute_auc(metabolism_df, col, individual_vars) %>% mutate(metric = col) } } }
auc_all <- bind_rows(auc_metab_list)
str(auc_all)tibble [66 × 6] (S3: tbl_df/tbl/data.frame) $ trace_id : chr [1:66] "1" "10" "11" "12" ... $ family_id_group: chr [1:66] "tweed" "blue" "tweed" "tweed" ... $ treatment_group: chr [1:66] "selected" "selected" "control" "selected" ... $ AUC : num [1:66] 0.412 0.245 0.164 0.743 0.458 ... $ n_timepoints : int [1:66] 5 5 5 5 5 5 5 5 5 5 ... $ metric : chr [1:66] "metabolism_per_area_mm2_measurement" "metabolism_per_area_mm2_measurement" "metabolism_per_area_mm2_measurement" "metabolism_per_area_mm2_measurement" ...AUC summary tables
sum_vars <- intersect( c(“metric”, “family_id_group”, “treatment_group”), names(auc_all) ) auc_summary <- auc_all %>% group_by(across(all_of(sum_vars))) %>% summarise( n = n(), mean = mean(AUC, na.rm = TRUE), sd = sd(AUC, na.rm = TRUE), se = sd / sqrt(n), median = median(AUC, na.rm = TRUE), .groups = “drop” )
print(auc_summary)# A tibble: 4 × 8 metric family_id_group treatment_group n mean sd se median <chr> <chr> <chr> <int> <dbl> <dbl> <dbl> <dbl> 1 metabolism_pe… blue control 17 0.346 0.203 0.0492 0.261 2 metabolism_pe… blue selected 16 0.316 0.191 0.0477 0.246 3 metabolism_pe… tweed control 17 0.331 0.189 0.0459 0.309 4 metabolism_pe… tweed selected 16 0.277 0.159 0.0398 0.221Statistical Analysis
Each individual clam (sample_id_group) is the observational unit. The model is built from whichever grouping factors are present: both family and treatment (with interaction) when both exist, or a one-way model when only one factor is available. Each plate maps to a unique family × treatment combination, so plate-level and group-level variance are confounded; interpret accordingly.
has_treatment <- “treatment_group” %in% names(auc_df) && length(unique(na.omit(auc_df\(treatment_group))) > 1 if (!has_family && !has_treatment) { return(list(model = NULL, anova = NULL, pairs_full = empty, pairs_family = empty, pairs_trt = empty, has_family = FALSE, has_treatment = FALSE)) } if (has_family) auc_df <- auc_df %>% mutate(family = factor(family_id_group)) if (has_treatment) auc_df <- auc_df %>% mutate(treatment = factor(treatment_group)) formula_str <- if (has_family && has_treatment) “AUC ~ family * treatment” else if (has_family) “AUC ~ family” else “AUC ~ treatment” model <- lm(as.formula(formula_str), data = auc_df) anova_res <- anova(model) if (has_family && has_treatment) { pairs_full <- as.data.frame(pairs(emmeans(model, ~ family * treatment), adjust = “tukey”)) pairs_family <- as.data.frame(pairs(emmeans(model, ~ family), adjust = “tukey”)) pairs_trt <- as.data.frame(pairs(emmeans(model, ~ treatment), adjust = “tukey”)) } else if (has_family) { pairs_family <- as.data.frame(pairs(emmeans(model, ~ family), adjust = “tukey”)) pairs_full <- pairs_family pairs_trt <- empty } else { pairs_trt <- as.data.frame(pairs(emmeans(model, ~ treatment), adjust = “tukey”)) pairs_full <- pairs_trt pairs_family <- empty } list( model = model, anova = anova_res, pairs_full = pairs_full, pairs_family = pairs_family, pairs_trt = pairs_trt, has_family = has_family, has_treatment = has_treatment ) } metrics_to_test <- unique(auc_all\)metric) stats_results <- map( set_names(metrics_to_test), ~ run_auc_stats(auc_all %>% filter(metric == .x)) ) ``` ## Results by metric
10.1.1 Metric: metabolism_per_area_mm2_measurement
ANOVA: Analysis of Variance Table
Response: AUC Df Sum Sq Mean Sq F value Pr(>F) family 1 0.01136 0.011360 0.3266 0.5697 treatment 1 0.02843 0.028429 0.8173 0.3695 family:treatment 1 0.00254 0.002541 0.0730 0.7879 Residuals 62 2.15653 0.034783
Pairwise: family × treatment (Tukey): contrast estimate SE df t.ratio p.value blue control - tweed control 0.01420026 0.06396940 62 0.222 0.9961 blue control - blue selected 0.02911310 0.06496123 62 0.448 0.9697 blue control - tweed selected 0.06814272 0.06496123 62 1.049 0.7214 tweed control - blue selected 0.01491284 0.06496123 62 0.230 0.9957 tweed control - tweed selected 0.05394246 0.06496123 62 0.830 0.8398 blue selected - tweed selected 0.03902962 0.06593814 62 0.592 0.9342
P value adjustment: tukey method for comparing a family of 4 estimates
Pairwise: family main effect: contrast estimate SE df t.ratio p.value blue - tweed 0.02661494 0.04593453 62 0.579 0.5644
Results are averaged over the levels of: treatment
Pairwise: treatment main effect: contrast estimate SE df t.ratio p.value control - selected 0.04152778 0.04593453 62 0.904 0.3695
Results are averaged over the levels of: family
11 AUC Box Plots with Statistical Annotations
Significance labels: *** p < 0.001, ** p < 0.01, * p < 0.05. Brackets are drawn only for significant pairs (p < 0.05). Plots are generated for whichever grouping factors are present: treatment-only, family-only, all-groups, within-family, and within-treatment.
sig_label <- function(p) {
case_when(p < 0.001 ~ "***", p < 0.01 ~ "**", p < 0.05 ~ "*",
TRUE ~ "ns")
}
# Add significance brackets to an existing ggplot.
# pairs_df : data frame with $contrast and $p.value columns
# group_levels: ordered character vector matching x-axis factor levels
# y_vals : numeric vector of AUC values used to set bracket heights
add_sig_brackets <- function(p, pairs_df, group_levels, y_vals) {
sig_pairs <- pairs_df %>%
mutate(label = sig_label(p.value)) %>%
filter(label != "ns")
if (nrow(sig_pairs) == 0) return(p)
y_max <- max(y_vals, na.rm = TRUE)
y_range <- diff(range(y_vals, na.rm = TRUE))
step <- y_range * 0.12
for (i in seq_len(nrow(sig_pairs))) {
parts <- str_split(as.character(sig_pairs$contrast[i]), " - ", 2)[[1]]
g1 <- trimws(parts[1])
g2 <- trimws(parts[2])
x1 <- match(g1, group_levels)
x2 <- match(g2, group_levels)
if (is.na(x1) || is.na(x2)) next
bar_y <- y_max + i * step
p <- p +
annotate("segment", x = x1, xend = x2,
y = bar_y, yend = bar_y,
colour = "black", linewidth = 0.6) +
annotate("segment", x = x1, xend = x1,
y = bar_y, yend = bar_y - step * 0.3,
colour = "black", linewidth = 0.6) +
annotate("segment", x = x2, xend = x2,
y = bar_y, yend = bar_y - step * 0.3,
colour = "black", linewidth = 0.6) +
annotate("text", x = (x1 + x2) / 2,
y = bar_y + step * 0.15,
label = sig_pairs$label[i], size = 4.5)
}
p
}for (met in metrics_to_test) {
df <- auc_all %>% filter(metric == met)
stats <- stats_results[[met]]
y_lab <- auc_y_label(met)
has_fam <- stats$has_family
has_trt <- stats$has_treatment
# ── Treatment main effect (x = treatment, tick = treatment name) ───────
if (has_trt) {
df_p <- df %>%
mutate(x = factor(treatment_group, levels = sort(unique(treatment_group))))
grps <- levels(df_p$x)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = trt_colours[grps], guide = "none") +
labs(x = "Treatment", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, stats$pairs_trt, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_treatment_", met, ".png")),
p, width = 5, height = 5)
}
# ── Family main effect (x = family, tick = family name) ───────────────
if (has_fam) {
df_p <- df %>%
mutate(x = factor(family_id_group, levels = sort(unique(family_id_group))))
grps <- levels(df_p$x)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fam_colours[grps], guide = "none") +
labs(x = "Family", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, stats$pairs_family, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_family_", met, ".png")),
p, width = 5, height = 5)
}
# Remaining plots require both factors
if (!has_fam || !has_trt) next
# ── All family:treatment groups (x = family:treatment) ─────────────────
# emmeans contrasts use spaces; convert to colon to match tick labels
pairs_fc <- stats$pairs_full %>%
mutate(contrast = str_replace_all(
contrast,
"([a-z]+) ([a-z]+)( - )([a-z]+) ([a-z]+)",
"\\1:\\2\\3\\4:\\5"
))
df_p <- df %>%
mutate(x = factor(
paste(family_id_group, treatment_group, sep = ":"),
levels = sort(unique(paste(family_id_group, treatment_group, sep = ":")))
))
grps <- levels(df_p$x)
fill_map <- setNames(make_palette(length(grps)), grps)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fill_map, guide = "none") +
labs(x = "Family : Treatment", y = y_lab) +
theme_classic(base_size = 13) +
theme(axis.text.x = element_text(angle = 20, hjust = 1))
p <- add_sig_brackets(p, pairs_fc, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_all_groups_", met, ".png")),
p, width = 6, height = 5)
# ── Within each family: treatment comparison (x = family:treatment) ────
# Tick labels are family:treatment so these plots are visually distinct
# from the treatment main-effect plot above.
for (fam in sort(unique(df$family_id_group))) {
df_p <- df %>%
filter(family_id_group == fam) %>%
mutate(x = factor(
paste(family_id_group, treatment_group, sep = ":"),
levels = sort(unique(paste(family_id_group, treatment_group, sep = ":")))
))
grps <- levels(df_p$x)
pairs_sub <- pairs_fc %>%
filter(str_count(contrast, paste0(fam, ":")) == 2)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fill_map[grps], guide = "none") +
labs(x = "Family : Treatment", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, pairs_sub, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_", fam, "_trt_", met, ".png")),
p, width = 5, height = 5)
}
# ── Within each treatment: family comparison (x = family:treatment) ────
# Tick labels are family:treatment so these plots are visually distinct
# from the family main-effect plot above.
for (trt in sort(unique(df$treatment_group))) {
df_p <- df %>%
filter(treatment_group == trt) %>%
mutate(x = factor(
paste(family_id_group, treatment_group, sep = ":"),
levels = sort(unique(paste(family_id_group, treatment_group, sep = ":")))
))
grps <- levels(df_p$x)
pairs_sub <- pairs_fc %>%
filter(str_count(contrast, paste0(":", trt)) == 2)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fill_map[grps], guide = "none") +
labs(x = "Family : Treatment", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, pairs_sub, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_", trt, "_fam_", met, ".png")),
p, width = 5, height = 5)
}
}
12 Save Output Data
write_csv(auc_all, file.path(out_dir, "auc_all_metrics.csv"))
write_csv(auc_summary, file.path(out_dir, "auc_summary.csv"))
if (nrow(metabolism_df) > 0)
write_csv(metabolism_df,
file.path(out_dir, "metabolism.csv"))
stats_compiled <- map_dfr(metrics_to_test, function(met) {
bind_rows(
stats_results[[met]]$pairs_full %>%
mutate(comparison = "family:treatment"),
stats_results[[met]]$pairs_family %>%
mutate(comparison = "family"),
stats_results[[met]]$pairs_trt %>%
mutate(comparison = "treatment")
) %>% mutate(metric = met)
})
write_csv(stats_compiled,
file.path(out_dir, "pairwise_stats.csv"))
message("Output files written to: ", out_dir)MATERIALS & METHODS
Control clams (C) compared with selected clams (S) at 36oC.
Clams were processed in 12-well plates and submerged in 5.0mL of resazurin working solution prepared today.
- 986.66 mL filtered seawater (4oC filtered sea water from 11/19/2025 by AH)
- 2.22 mL resazurin stock solution (from step 1 above)
- 1.00 mL DMSO
- 10.00 mL antibiotic solution (100x Penn/Strep & 100x Fungizone)
Plates were measured every 30mins on a Synergy HTX (Agilent) plate reader over the course of 4hrs.
Plate layout was randomized; Steven is currently the only person who knows the treatment assignments.
Low volumes were noted in the following plates/wells, which may have been due to clam spitting:
DA1 BA3 DB2 EC2
Plate temps
Wells were spot checked and range noted in table below.
| PLATE | TIME | TEMP |
|---|---|---|
| A | 0 | 11-12 |
| A | 1 | 29 |
| A | 2 | 31 |
| A | 3 | 32-33 |
| A | 4 | 33-34 |
RESULTS
All six plates passed the consistency check (12 wells per plate at every timepoint). Although four wells were flagged for low volume in the experimental notes (D A1, B A3, D B2, E C2), they were not marked for exclusion in the layout file, so no wells were removed from analysis. Notably, Plate A experienced a slow warm-up, reaching only 11–12°C at T0 and 33–34°C by T4; this thermal lag should be considered when interpreting Plate A data.
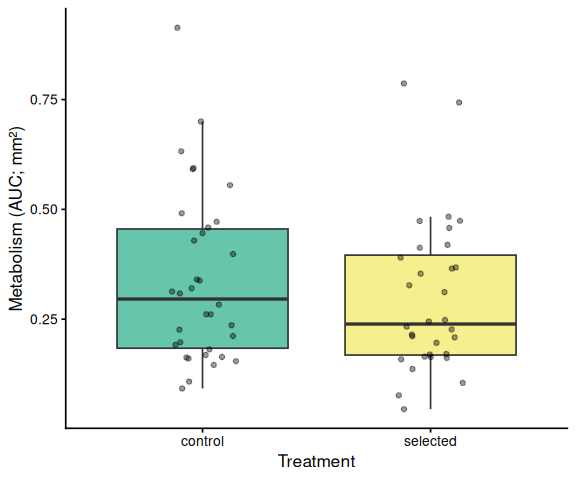
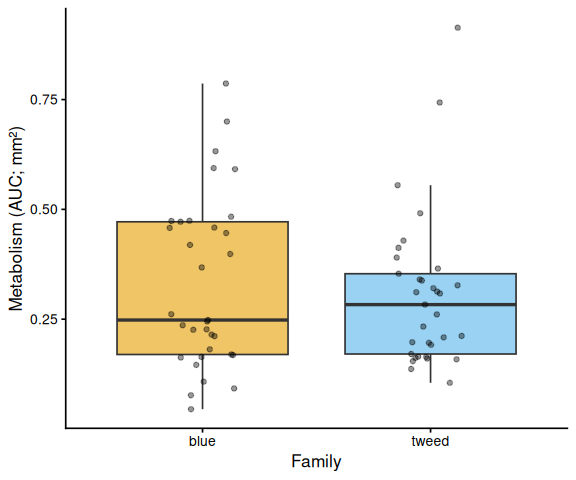
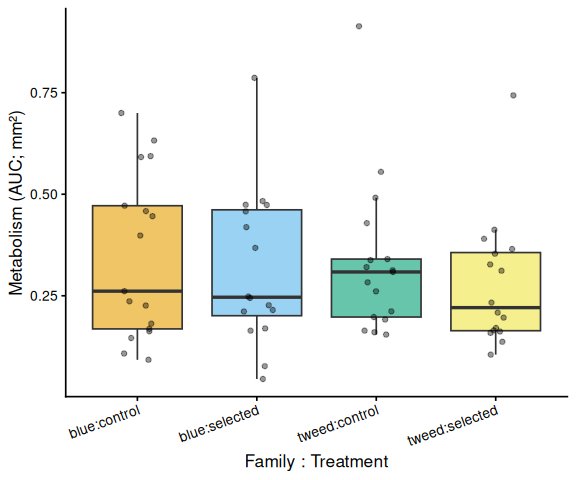
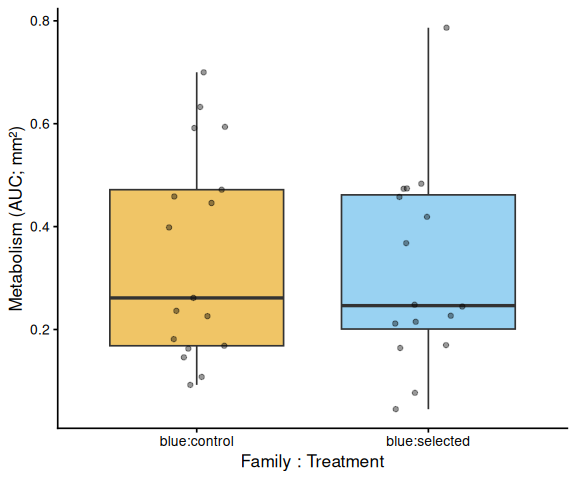
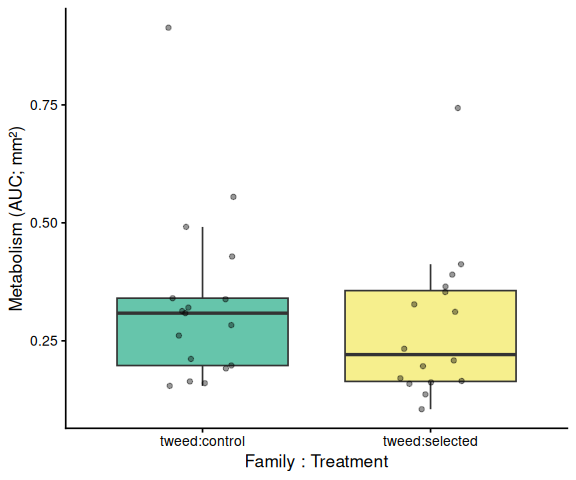
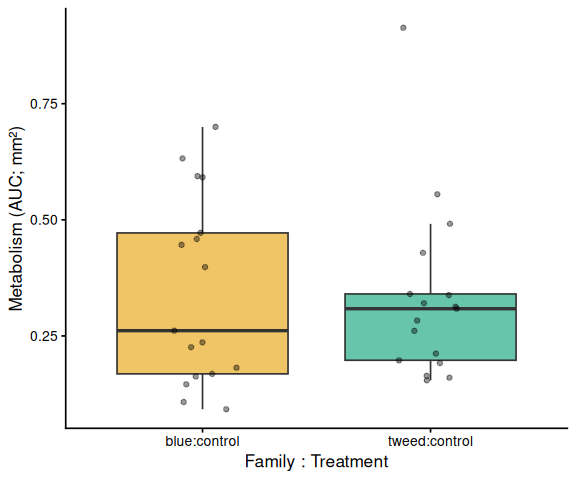
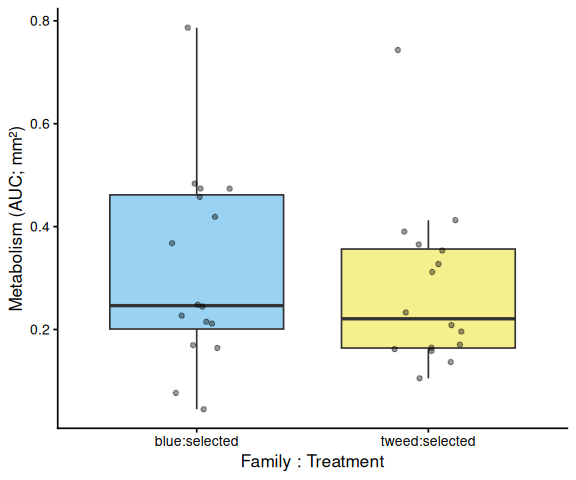
Metabolic activity (AUC of blank-corrected, size-normalised fold-change per mm² shell area) showed no significant effects of family, treatment, or their interaction (ANOVA: family F = 0.33, p = 0.570; treatment F = 0.82, p = 0.370; family × treatment F = 0.07, p = 0.788). Group mean AUCs were similar across all four groups: blue control (0.346 ± 0.049 SE), tweed control (0.331 ± 0.046), blue selected (0.316 ± 0.048), and tweed selected (0.277 ± 0.040). No pairwise contrast reached significance (all Tukey-adjusted p > 0.33).
The time-series mixed-effects model likewise detected no significant pairwise differences between groups at any timepoint (all Tukey-adjusted p > 0.14).
In summary, no metabolic differences were detected between selected and control clams, or between families, on Day 1 of the repeated 36°C heat stress experiment. The absence of the blue-control elevation seen on Day 0 may reflect either recovery effects from overnight cold storage or the thermal lag of Plate A during this assay.