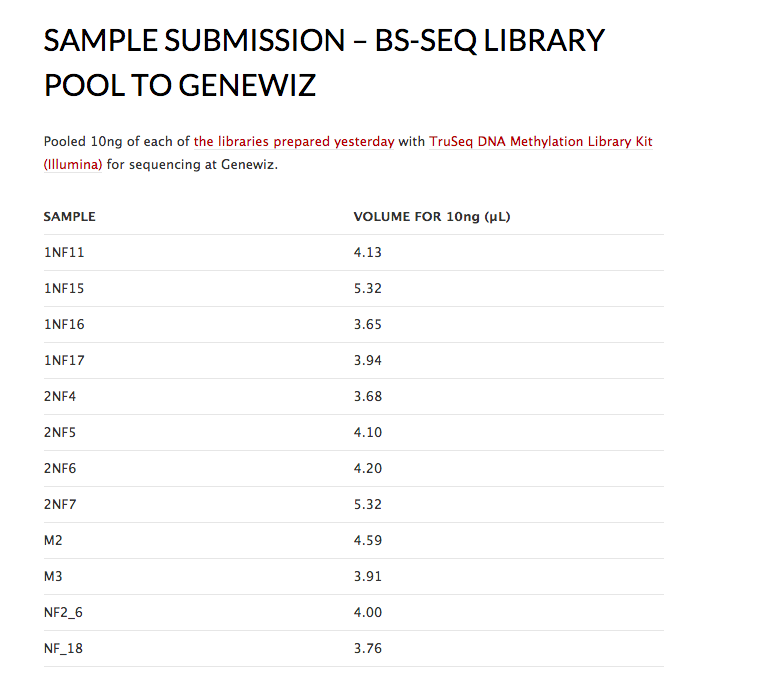
For the 12 samples
Select 4 samples from 1NF gel take 2
Select 4 samples from 2NF gel take 2
Select 2 from gel take 2 Lotterhos
M1
M2
M3
Select 2 from the following sent to Katie (do not have to run on gel)
NF2 14
NF2 6
NF2 18
NF2 15
NF2 17

Short term will just check out the first 8.¶
These are samples outplanted at Oyster Bay and Fidalgo, and in both cases parents from Fidalgo.
The hypothesis is that Epigenetic pattern will differ (and we can attribute to Environment)
Quick look at raw data¶

Sequencing Platform: Illumina HiSeq2500
Read Type/Length: 50bp single-end, single index
Total Number of Reads: 116,280,817
Reads Per File:
11_GGCTAC_L001_R1_001.fastq.gz 10933121
12_CTTGTA_L001_R1_001.fastq.gz 10816647
1_ATCACG_L001_R1_001.fastq.gz 9402890
2_CGATGT_L001_R1_001.fastq.gz 11954873
3_TTAGGC_L001_R1_001.fastq.gz 11817358
4_TGACCA_L001_R1_001.fastq.gz 11606618
5_ACAGTG_L001_R1_001.fastq.gz 12589609
6_GCCAAT_L001_R1_001.fastq.gz 12489766
7_CAGATC_L001_R1_001.fastq.gz 10295293
8_ACTTGA_L001_R1_001.fastq.gz 14374642Unzip¶
In [1]:
cd /Volumes/Histidine/hectocotylus/whole-BS-01
In [2]:
%%bash
for f in *.gz
do
STEM=$(basename "${f}" .gz)
gunzip -c "${f}" > /Volumes/Histidine/hectocotylus/whole-BS-01/fq/"${STEM}"
done
FastQC¶
In [3]:
!/Applications/bioinfo/FastQC/fastqc \
-o /Volumes/Histidine/hectocotylus/whole-BS-01/fq/ \
-t 4 \
/Volumes/Histidine/hectocotylus/whole-BS-01/fq/*

this unusual pattern seem to hold true..
In [ ]:
Recent Comments