Our current system for identifying differentially expressed transcripts relies on using the EdgeR Bioconductor package. We have a protocol and scripts described below for identifying differentially expressed transcripts and clustering transcripts according to expression profiles. This process is somewhat interactive, and described are automated approaches as well as manual approaches to refining gene clusters and examining their corresponding expression patterns.
|
Note
|
We recommend generating a single Trinity assembly based on combining all reads across all samples as inputs. Then, reads are aligned separately back to the single Trinity assembly for downstream analyses of differential expression, according to our abundance estimation protocol. If you decide to assemble each sample separately, then you’ll likely have difficulty comparing the results across the different samples due to differences in assembled transcript lengths and contiguity. |
|
Note
|
If you have biological replicates, align each replicate set of reads and estimate abundance values for the Trinity contigs independently. |
First, join the RSEM-estimated abundance values for each of your samples by running:
$TRINITY_HOME/util/RSEM_util/merge_RSEM_frag_counts_single_table.pl sampleA.RSEM.isoform.results sampleB.RSEM.isoform.results ... > transcripts.counts.matrixDo the same for the RSEM gene files (where counts are aggregated per Trinity component) like so:
$TRINITY_HOME/util/RSEM_util/merge_RSEM_frag_counts_single_table.pl sampleA.RSEM.gene.results sampleB.RSEM.gene.results ... > genes.counts.matrixEdit the column headers in each of the matrix files to your liking, since this is how the samples (and replicates) will be named in the downstream analysis steps.
Identifying Differentially Expressed Transcripts
Trinity currently supports the use of Bioconductor tools (edgeR and DESeq) for differential expression analysis. To perform the steps below, you must have the R software installed along with certain libraries and packages shown below. To install Bioconductor and the required packages, run the following from an R prompt:
source("http://bioconductor.org/biocLite.R")
biocLite('edgeR')
biocLite('DESeq')
biocLite('ctc')
biocLite('Biobase')
install.packages('gplots’)
install.packages(‘ape’)Differentially expressed transcripts or genes are identified by running the script below, which will perform pairwise comparisons among each of your sample types. If you have biological replicates for each sample, you should indicate this as well (described further below). To analyze transcripts, use the transcripts.counts.matrix file. To perform an analysis at the gene level, use the genes.counts.matrix. Again, Trinity Components are used as a proxy for gene level studies.
$TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl#################################################################################################
#
# Required:
#
# --matrix <string> matrix of raw read counts (not normalized!)
#
# --method <string> edgeR|DESeq (DESeq only supported here w/ bio replicates)
#
#
# Optional:
#
# --samples_file <string> tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1
# cond_A cond_A_rep2
# cond_B cond_B_rep1
# cond_B cond_B_rep2
#
#
# General options:
#
# --min_rowSum_counts <int> default: 10 (only those rows of matrix meeting requirement will be tested)
#
# --output|o aname of directory to place outputs (default: $method.$pid.dir)
#
###############################################################################################
#
# ## EdgeR-related parameters
# ## (no biological replicates)
#
# --dispersion <float> edgeR dispersion value (default: 0.1) set to 0 for poisson (sometimes breaks...)
#
# http://www.bioconductor.org/packages/release/bioc/html/edgeR.html
#
###############################################################################################
#
# ## DE-Seq related parameters
#
# --DESEQ_method <string> "pooled", "pooled-CR", "per-condition", "blind"
# --DESEQ_sharingMode <string> "maximum", "fit-only", "gene-est-only"
# --DESEQ_fitType <string> fitType = c("parametric", "local")
#
# ## (no biological replicates)
# note: FIXED as: method=blind, sharingMode=fit-only
#
# http://www.bioconductor.org/packages/release/bioc/html/DESeq.html
#
################################################################################################|
Note
|
Based on our experiences with differential expression analysis and Trinity assemblies, we currently endorse edgeR as our method of choice. |
Identifying DE features: No biological replicates
Using edgeR without replicates:
Analysis/DifferentialExpression/run_DE_analysis.pl --matrix counts.matrix --method edgeRIdentifying DE features: With biological replicates (PREFERRED)
Be sure to have a samples_described.txt file that describes the relationship between samples and replicates. For example:
conditionA condA-rep1
conditionA condA-rep2conditionB condB-rep1
conditionB condB-rep2conditionC condC-rep1
conditionC condC-rep2where condA-rep1, condA-rep2, condB-rep1, etc…, are all column names in the counts.matrix generated earlier (see top of page). Your sample names that group the replicates are user-defined here.
Using edgeR with replicates:
$TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl --matrix SP2.rnaseq.counts.matrix --method edgeR --samples_file samples_described.txt|
Note
|
A full example of the edgeR pipeline involving combining reads from multiple samples, assembling them using Trinity, separately aligning reads back to the trintiy assemblies, abundance estimation using RSEM, and differential expression analysis using edgeR is provided at: $TRINITY_HOME/sample_data/test_full_edgeR_pipeline |
Analyzing The Differentially Expressed Transcripts
Computing normalized expression values
The above differential expression studies are based on statistics that leverage the raw counts. For studies beyond identifying statistically significant differences in gene expression for individual genes or transcripts between samples, we need to compute the expression values for these features. The number of reads observed for a given feature depends on several factors including the depth of sequencing, the length of the feature (longer transcripts generate more fragment reads), and the expression level of the feature. Expression values normalized for each of these factors are measured in FPKM (fragments per feature kilobase per million reads mapped). The term feature is used here because it could be a transcript, a gene, an exon, or just about anything that can be quantified as a mappable unit. Also, before computing FPKM values, we must normalize the RNA-Seq count data to make them comparable across the muliple samples and replicates. We first use TMM normalization to account for differences in the mass composition of the RNA-seq samples, which doesn’t change the fragment count data, but instead provides a scaling parameter that yields an effective library size (total mappable reads) for each sample. This effective library size is then used in the FPKM calculations.
Compute the TMM-normalized FPKM values for all samples like so:
First, extract the transcript length information from one of the RSEM output files like so:
cat sampleA.RSEM.isoforms.results | cut -f1,3,4 > feature_lengths.txtUsing the counts.matrix file created above, perform TMM normalization and generate the FPKM values per transcript and sample as follows:
$TRINITY_HOME/Analysis/DifferentialExpression/run_TMM_normalization_write_FPKM_matrix.pl --matrix counts.matrix --lengths feature_lengths.txtThe above should generate a file: counts.matrix.TMM_normalized.FPKM (adding a suffix to your --matrix filename), which can be used for downstream analyses such as for clustering transcripts based on expression profiles and generating heatmaps (see below).
Extracting and clustering differentially expressed transcripts
An initial step in analyzing differential expression is to extract those transcripts that are most differentially expressed (most significant P-values and fold-changes) and to cluster the transcripts according to their patterns of differential expression across the samples. To do this, you can run the following from within the edgeR output directory, bu running the following script:
$TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl####################################################################################
#
# Required:
#
# --matrix matrix.normalized.FPKM
#
# Optional:
#
# -P p-value cutoff for FDR (default: 0.001)
#
# -C min abs(log2(a/b)) fold change (default: 2 (meaning 2^(2) or 4-fold).
#
# --output prefix for output file (default: "diffExpr.P${Pvalue}_C${C})
#
# Clustering methods:
#
# --gene_dist <string> euclidean, pearson, spearman, (default: euclidean)
# maximum, manhattan, canberra, binary, minkowski
#
# --gene_clust <string> ward, single, complete, average, mcquitty, median, centroid (default: complete)
#
#
#
####################################################################################For example:
$TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl --matrix matrix.TMM_normalized.FPKM -P 1e-3 -C 2
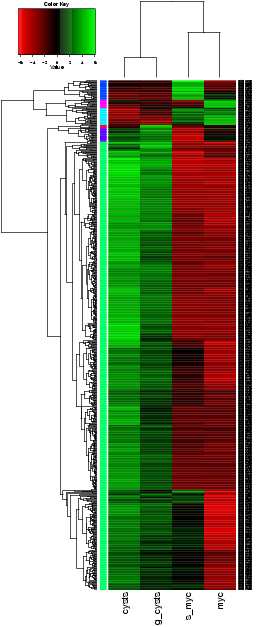
The above is mostly just a visual reference. To more seriously study and define your gene clusters, you will need to interact with the data as described below. The clusters and all required data for interrogating and defining clusters is all saved with an R-session, locally with the file all.RData. This will be leveraged as described below.
Automatically defining a K-number of Gene Clusters
Run the script below to automatically split the data set into a sets of transcripts with related expression patterns by partitioning the hierarchically clustered transcript tree.
$TRINITY_HOME/Analysis/DifferentialExpression/define_clusters_by_cutting_tree.pl###################################################################################
#
# -K <int> define K clusters via k-means algorithm
#
# or, cut the hierarchical tree:
#
# --Ktree <int> cut tree into K clusters
#
# --Ptree <float> cut tree based on this percent of max(height) of tree
#
# -R <string> the filename for the store RData (file.all.RData)
#
###################################################################################For example:
$TRINITY_HOME/Analysis/DifferentialExpression/define_clusters_by_cutting_tree.pl --Ptree $percent_tree_heightA directory will be created called: diffExpr.P0.001_C2.matrix.R.all.RData.clusters_fixed_P_20/ and contain the expression matrix for each of the clusters (log2-transformed, median centered).
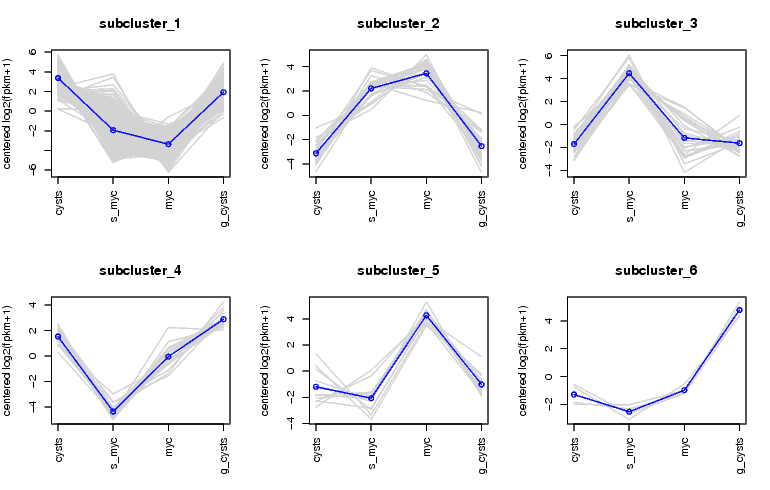
To plot the mean-centered expression patterns for each cluster, visit that directory and run:
$TRINITY_HOME/Analysis/DifferentialExpression/plot_expression_patterns.pl subcluster_*This will generate a summary image file: my_cluster_plots.pdf, as shown below:
Manually Defining Gene Clusters
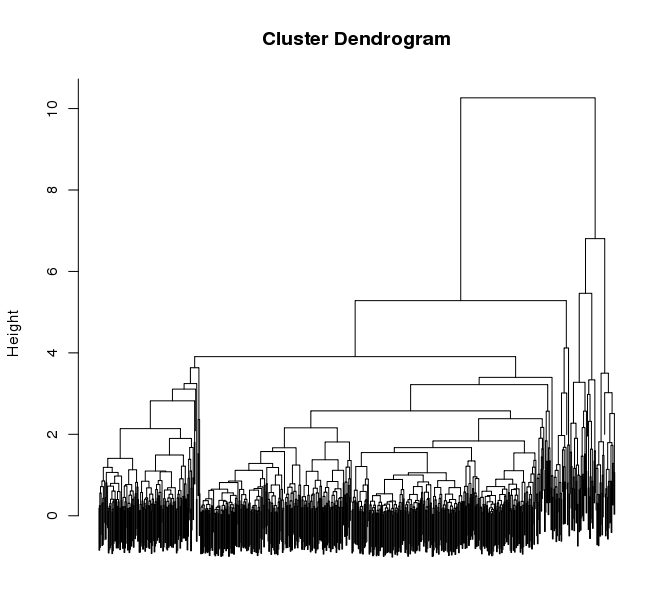
Manually defining your clusters is the best way to organize the data to your liking. This is an interactive process. Fire up R from within your output directory, being sure it contains the all.RData file, and enter the following commands:
% R> load("all.RData") # check for your corresponding .RData file name to use here, replace all.RData accordingly> source("$TRINITY_HOME/Analysis/DifferentialExpression/R/manually_define_clusters.R")> manually_define_clusters(hc_genes, centered_data)This should yield a display containing the hierarchically clustered genes, as shown below:
Now, manually define your clusters from left to right (order matters here, so you can decipher the results later!) by clicking on the branch vertical branch that defines the clade of interest. After clicking on the branch, it will be drawn with a red box around the selected clade, as shown below:
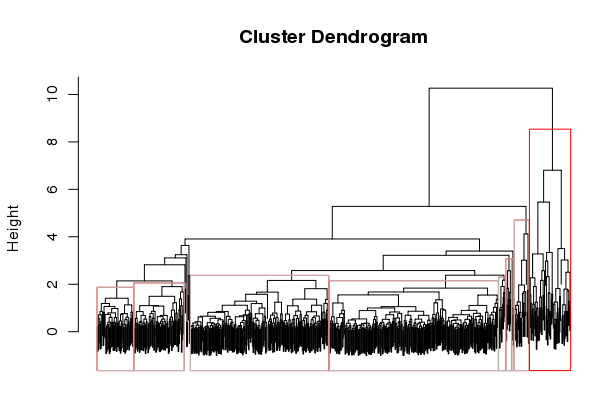
Right click with the mouse (or double-touch a touchpad) to exit from cluster selection.
The clusters as selected will be written to a subdirectory manually_defined_clusters_$count_clusters, and exist in a format similar to the automated-selection of clusters described above. Likewise, you can generate plots of the expression patterns for each cluster using the plot_expression_patterns.pl script.