Read Alignment
Our standard practice for aligning reads to the Trinity transcripts for visualization and quality assessment is to run alignReads.pl with the --bowtie option, like so:
TRINITY_RNASEQ_ROOT/util/alignReads.pl --left left.fq --right right.fq --seqType fq \
--target Trinity.fasta --aligner bowtie --retain_intermediate_files|
Note
|
If your data are strand-specific, be sure to set --SS_lib_type as done with Trinity.pl |
This alignment process generates lots of output files, ex. for paired strand-specific data:
-rw-rw-r-- 1 bhaas broad 33644239 Oct 29 09:51 bowtie_out.coordSorted.sam
-rw-rw-r-- 1 bhaas broad 5761928 Oct 29 09:51 bowtie_out.coordSorted.bam
-rw-rw-r-- 1 bhaas broad 33644239 Oct 29 09:51 bowtie_out.nameSorted.sam
-rw-rw-r-- 1 bhaas broad 4256476 Oct 29 09:51 bowtie_out.nameSorted.bam
-rw-rw-r-- 1 bhaas broad 4416 Oct 29 09:51 bowtie_out.coordSorted.bam.bai
-rw-rw-r-- 1 bhaas broad 33634652 Oct 29 09:51 bowtie_out.coordSorted.sam.+.sam
-rw-rw-r-- 1 bhaas broad 9587 Oct 29 09:51 bowtie_out.coordSorted.sam.-.sam
-rw-rw-r-- 1 bhaas broad 33634652 Oct 29 09:51 bowtie_out.nameSorted.sam.+.sam
-rw-rw-r-- 1 bhaas broad 9587 Oct 29 09:51 bowtie_out.nameSorted.sam.-.sam
-rw-rw-r-- 1 bhaas broad 5759999 Oct 29 09:51 bowtie_out.coordSorted.sam.+.bam
-rw-rw-r-- 1 bhaas broad 4416 Oct 29 09:51 bowtie_out.coordSorted.sam.+.bam.bai
-rw-rw-r-- 1 bhaas broad 1836 Oct 29 09:51 bowtie_out.coordSorted.sam.-.bam
-rw-rw-r-- 1 bhaas broad 1680 Oct 29 09:51 bowtie_out.coordSorted.sam.-.bam.bai
-rw-rw-r-- 1 bhaas broad 4255371 Oct 29 09:51 bowtie_out.nameSorted.sam.+.bam
-rw-rw-r-- 1 bhaas broad 1843 Oct 29 09:51 bowtie_out.nameSorted.sam.-.bamIf you do not have strand-specific reads, then you’ll not have the (+) and (-) versions of the files as above.
The bowtie_out.coordSorted.bam file contains both properly-mapped pairs and single unpaired fragment reads. This file can be used for visualizing the alignments and coverage data using IGV (below).
You can examine the name-sorted file to assess the numbers of distinct reads that are found to align to transcripts as properly paired, individually, or as improper pairs. Proper pairing requires that the paired fragment reads point towards each other and are within the maximum specified fragment size (see alignReads.pl options via -h for more info).
Run the following script on the name-sorted sam file to obtain read alignment statistics:
% $TRINITY_HOME/util/SAM_nameSorted_to_uniq_count_stats.pl bowtie_out.nameSorted.sam.+.sam#read_type count pct
proper_pairs 21194964 93.22 :both read pairs align to a single contig and point toward each other.
left_only 836213 3.68 :only the left (/1) read is reported in an alignment
right_only 687576 3.02 :only the right (/2) read is reported in an alignment
improper_pairs 16640 0.07 :both leftr and right reads align, but to separate contigs, or to a single contig in the wrong expected relative orientations.Visualization
The Trinity Transcripts and read alignments can be visualized using the Integrated Genomics Viewer.
Just import the Trinity.fasta file as a genome, and load up the bam file containing the aligned reads. A screenshot below shows how the data are displayed:
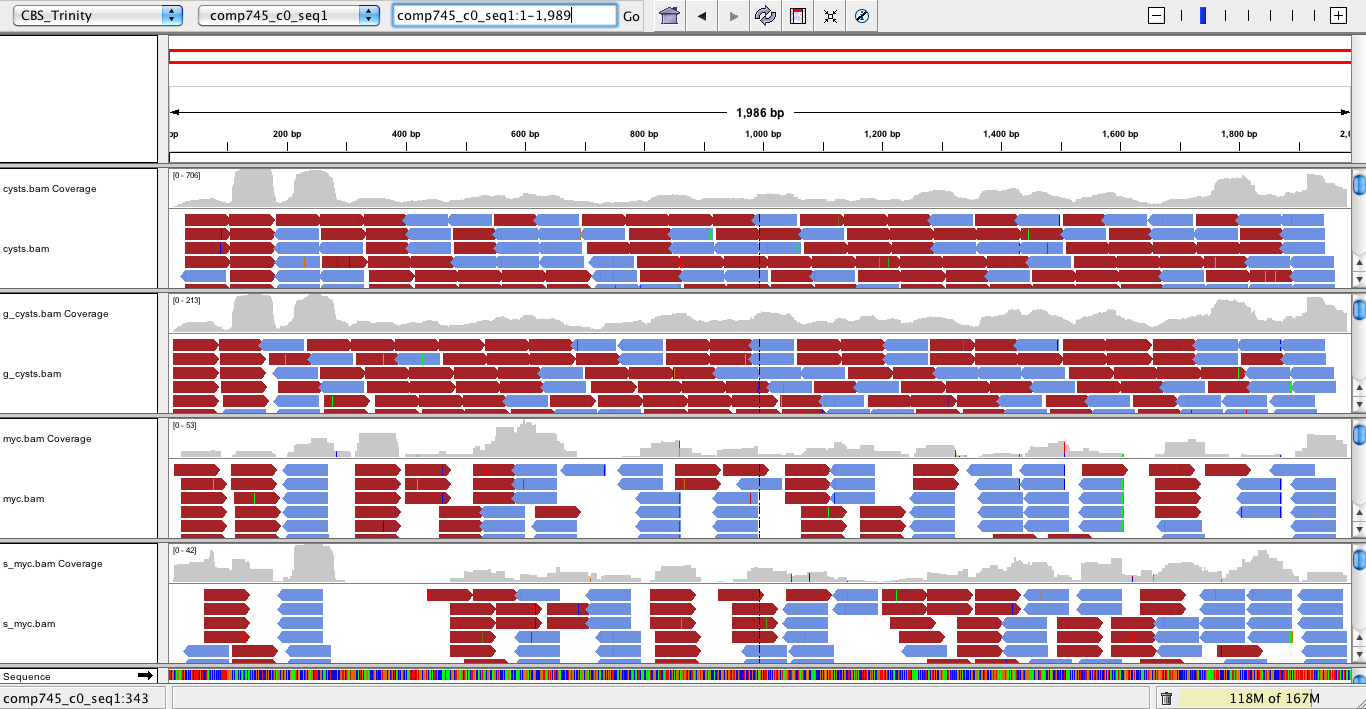
|
Note
|
If strand-specific RNA-Seq is used, each of the strand-specific bam files above can be loaded in as separate tiers. This can be useful for examining the evidence for sense and antisense transcription. Note that strand-specific RNA-Seq methods can still generate about 1% of background that will show up as antisense resulting from an artifact of the experimental method. |