RNA Isolation Protocol.
- 100 mg tissue homogenized in 1 ml RNAzol RT at room temp.
- Added 400 ul 0.1% DEPC water to homogenate.
- Incubated 15 minutes at room temp
- Centrifuged 15 minutes at 11,400 rpm (~12,000 g)
- Transferred 1 ml supernatant to fresh tube
- Added 400 ul 75% EtOH
- Mixed via inversion for 15 seconds
- Incubated samples 10 minutes at room temp
- Centrifuged 8 minutes at 11,400 rpm (~12,000 g)
- Discarded supernant
- Added 600 ul 75% EtOH
- Vortexed 15 seconds
- Centrifuged 3 minutes at 7,400 rpm (~8000 g)
- Repeated steps 10 - 13.
- Removed supernatant
- resuspended RNA in 50 ul 0.1% DEPC water
Instead of nanodropping the RNA, I immediately Turbo DNase Treated the samples to remove any DNA contamination.
DNase Treatment Protocol:
1. Added 5 ul DNase 10X Buffer to each 50 ul of RNA.
2. Added 1 ul Turbo DNase to each RNA.
3. Incubated at room temp for 30 minutes
4. Added 5 ul DNase inhibitor to each sample
5. Incubated at room temp for 5 minutes, flicking occasionally
6. Centrifuged at 9400 rpm (~10,000 g) for 1.5 minutes
7. Decanted the top 50 ul of supernatant to fresh tube
2. Added 1 ul Turbo DNase to each RNA.
3. Incubated at room temp for 30 minutes
4. Added 5 ul DNase inhibitor to each sample
5. Incubated at room temp for 5 minutes, flicking occasionally
6. Centrifuged at 9400 rpm (~10,000 g) for 1.5 minutes
7. Decanted the top 50 ul of supernatant to fresh tube
8. Nanodropped
Nanodrop Results (including positive control):
| Sample ID | Date | Time | ng/ul | A260 | A280 | 260/280 | 260/230 |
| C+ | 5/5/2015 | 4:17 PM | 167.3 | 4.182 | 2.092 | 2 | 2.34 |
| 42715HT1 | 5/5/2015 | 4:18 PM | 238.58 | 5.965 | 3.366 | 1.77 | 0.66 |
| 42715HT2 | 5/5/2015 | 4:19 PM | 422.45 | 10.561 | 5.431 | 1.94 | 1 |
| 42715HT3 | 5/5/2015 | 4:21 PM | 175.57 | 4.389 | 2.26 | 1.94 | 1.79 |
| 42715HT4 | 5/5/2015 | 4:22 PM | 251.19 | 6.28 | 3.532 | 1.78 | 0.68 |
| 42715HT5 | 5/5/2015 | 4:23 PM | 391.29 | 9.782 | 5.133 | 1.91 | 0.87 |
| 42715ST1 | 5/5/2015 | 4:24 PM | 690.94 | 17.274 | 10.258 | 1.68 | 0.49 |
| 42715ST2 | 5/5/2015 | 4:25 PM | 393.97 | 9.849 | 5.181 | 1.9 | 0.91 |
| 42715ST3 | 5/5/2015 | 4:25 PM | 365.73 | 9.143 | 4.917 | 1.86 | 0.99 |
| 42715ST4 | 5/5/2015 | 4:26 PM | 426.33 | 10.658 | 5.407 | 1.97 | 0.99 |
| 42715ST5 | 5/5/2015 | 4:27 PM | 343.49 | 8.587 | 4.622 | 1.86 | 0.81 |
For the qPCR I used Actin primers and a positive control from Fidalgo seed oysters extracted on 3/23/2015. My negative control contained no DNA as to show there was no contamination in the Master Mix. I also bumped up the volume of sample used to 0.5 ul due to issues with the pipetter not being able to accurately pipette 0.2 ul. This is about 2.5 X the concentration of contamination gDNA that the cDNA will have in it. I also bumped down the amount of water by 1 ul per reaction so as to ensure proper reaction mixture ratios were maintained.
qPCR Reagent Table:
| Volume | Reactions X12 | |
| Ssofast Evagreen MM | 10 | 140 |
| FWD Primer | 0.5 | 7 |
| REV Primer | 0.5 | 7 |
| Nuclease Free H2O | 8.8 | 123.2 |
| RNA | 0.5 |
qPCR protocol:
1. Added each from greatest volume to least to make the master mix.
2. Pipetted 20 ul master mix into each well of a qPCR partial plate
3. Added 0.5 ul sample to each tube
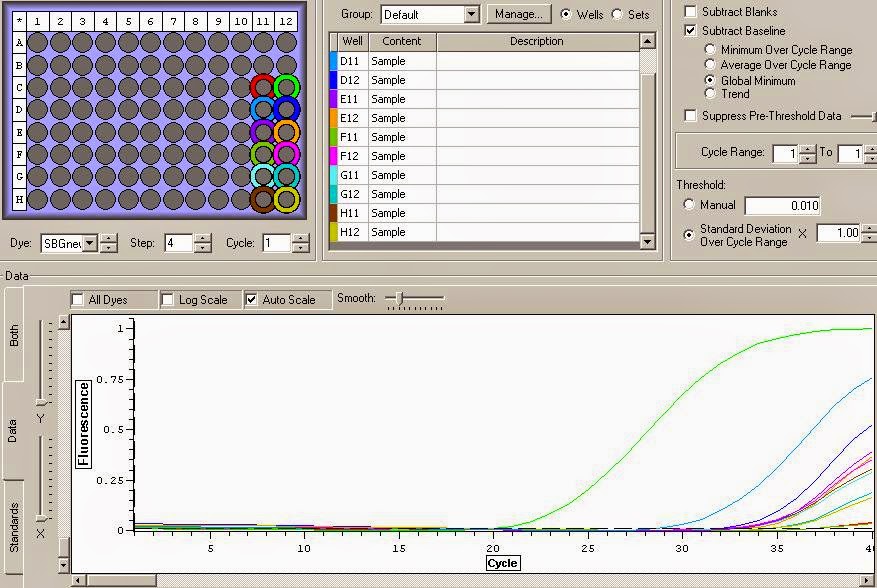
qPCR plate lay out:
| 11 | 12 | |
| C | C- | C+ |
| D | 42715HT1 | 42715ST1 |
| E | 42715HT2 | 42715ST2 |
| F | 42715HT3 | 42715ST3 |
| G | 42715HT4 | 42715ST4 |
| H | 42715HT5 | 42715ST5 |
This layout can be seen in the top left corner of the qPCR image below.
qPCR program:
| Sybr New Plate+Sybr cDNA 55 melt 1 Read | ||
| Step | Temperature | Time |
| Initiation | 95 C | 10 min |
| Elongation | 95 C | 15 sec |
| 55 C | 15 sec | |
| Read | ||
| 72 C | 15 sec | |
| Repeat Elongation 40 times | ||
| Termination | 95 C | 1 min |
| 55 C | 1 sec | |
| Melt Curve Manual ramp 0.2C per sec Read 0.5 C | 65 - 95 C | 30 sec |
| 21 C | 10 min | |
| End |
Results:
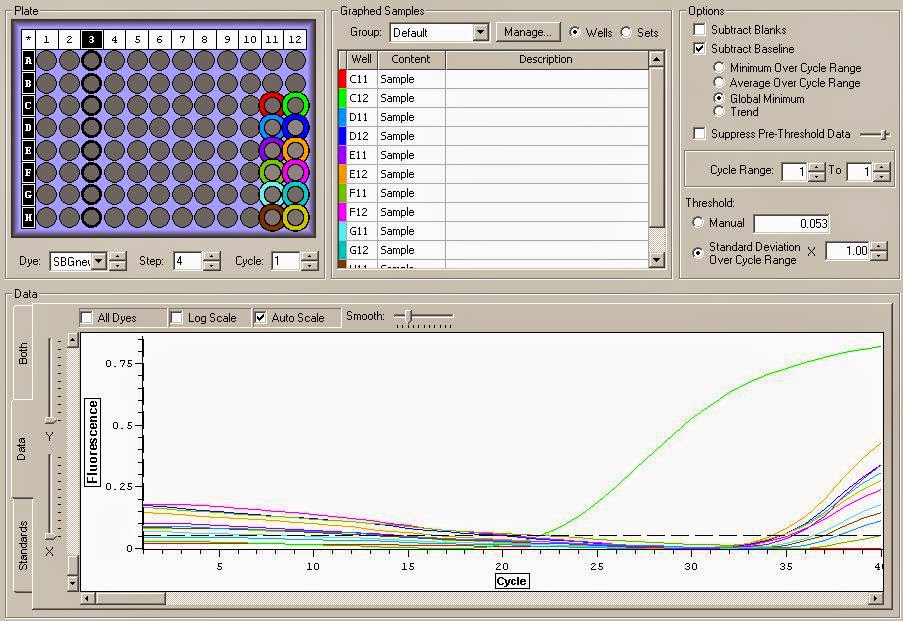
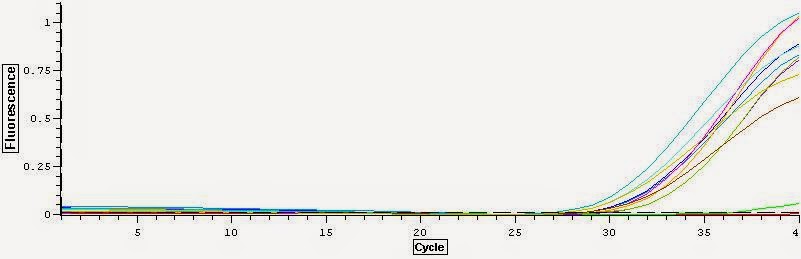
It looks like the DNase treatment removed some but not all the DNA. The DNA replicates much much earlier in the cycles. Overall I think these samples are good samples with high concentrations and mild DNA contamination. You can see the data files for the qPCR here and here.
Sometime this week I will begin the process of creating cDNA to use with qPCR to test for HSP70 and other RNA markers.