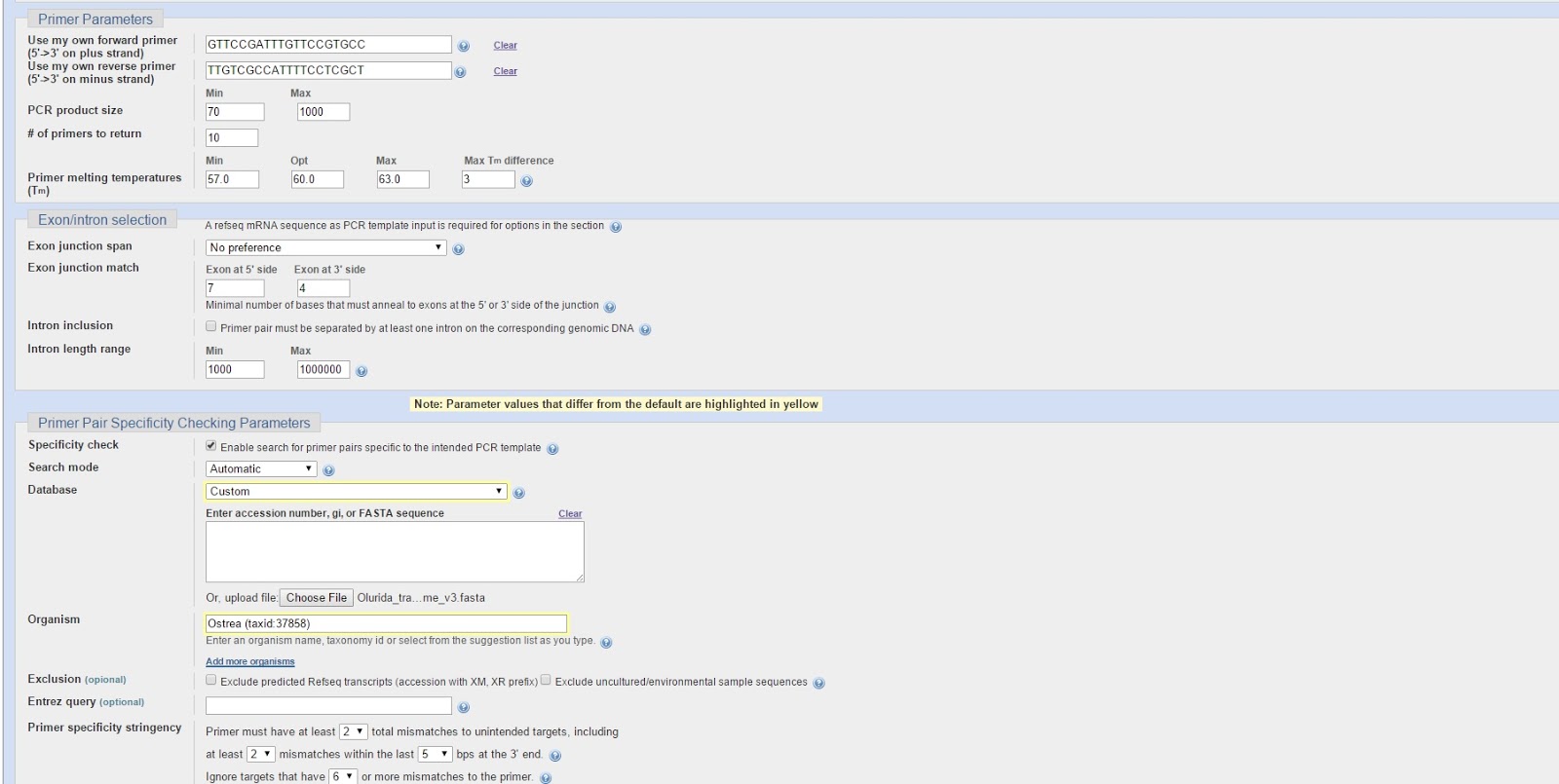
Primers
| 1672 | Flk_PGRP_FWD | AGCTGGTGCAGTCCTATCAG | JH | 6/1/2015 | 20 | 59 | O.lurida | PGRP-S Flanking | O75594 | |
| 1671 | Flk_PGRP_REV | TGTGTATGAAAAGTAATGAAGAGCA | JH | 6/1/2015 | 25 | 57 | O.lurida | O75594 | ||
| 1670 | Flk_CARM_FWD | TTCACACAGCCCATTGTGGAT | JH | 6/1/2015 | 21 | 60 | O.lurida | CARM1 Flanking | Q6DC04 | |
| 1669 | Flk_CARM_REV | TGGGATGGGTCAGATAAACCT | JH | 6/1/2015 | 21 | 58 | O.lurida | Q6DC04 | ||
| 1668 | Flk_BMP2_FWD | GGCTGGCTGGATCGTCAT | JH | 6/1/2015 | 18 | 60 | O.lurida | BMP2 Flanking | P12643 | |
| 1667 | Flk_BMP2_REV | ATGGAGTCTGTGGACGGTTTG | JH | 6/1/2015 | 21 | 60 | O.lurida | P12643 | ||
| 1666 | Flk_HSPb11_FWD | AGAATTGTCTGTGGAATCGAGC | JH | 6/1/2015 | 22 | 59 | O.lurida | HSPb11 Flanking | Q9Y547 | |
| 1665 | Flk_HSPb11_REV | ATCAACGCCAGGGGAACTTG | JH | 6/1/2015 | 20 | 61 | O.lurida | Q9Y547 | ||
| 1664 | Flk_PGE/EP4_FWD | GCTCAACGAATTGCTCTACTCC | JH | 6/1/2015 | 22 | 59 | O.lurida | PGE/EP4 Flanking | P32240 | |
| 1663 | Flk_PGE/EP4_REV | TCCGTCTGCTTTTTAGAATGGTA | JH | 6/1/2015 | 23 | 58 | O.lurida | P32240 | ||
| 1662 | Flk_GABABR_FWD | GAGGAGGACACGAAACTCCG | JH | 6/1/2015 | 20 | 60 | O.lurida | GABABR Flanking | Q9WV18 | |
| 1661 | Flk_GABABR_REV | TGCACCACACTCCTGATGAC | JH | 6/1/2015 | 20 | 60 | O.lurida | Q9WV18 | ||
| 1660 | Flk_GRB2_FWD | TCAGAACTGGTTCAAAGCTGAGT | JH | 6/1/2015 | 23 | 60 | O.lurida | GRB2 Flanking | P62994 | |
| 1659 | Flk_GRB2_REV | ACTGCGCTGACATACTGGAC | JH | 6/1/2015 | 20 | 60 | O.lurida | P62994 | ||
| 1658 | Flk_H3.3_FWD | CCAATGACAAATGAGCCACACAA | JH | 6/1/2015 | 23 | 60 | O.lurida | H3.3 Flanking | Q6P823 | |
| 1657 | Flk_H3.3_REV | TCGTACAAAGCAAACTGCACG | JH | 6/1/2015 | 21 | 60 | O.lurida | Q6P823 | ||
| 1656 | Flk_H2A.V_FWD | GCGATGGAGTTGATGAGGTG | JH | 6/1/2015 | 20 | 59 | O.lurida | H2A.V Flanking | P08991 | |
| 1655 | Flk_H2A.V_REV | CAAGGCAGTTTCTCGTTCGG | JH | 6/1/2015 | 20 | 59 | O.lurida | P08991 | ||
| 1654 | Flk_H2A_FWD | TGCTGGGGTTTTTCTGGGTC | JH | 6/1/2015 | 20 | 60 | O.lurida | H2A Flanking | P02270 | |
| 1653 | Flk_H2A_REV | TCAGGACGTGGTAAAGGAGGA | JH | 6/1/2015 | 21 | 60 | O.lurida | P02270 | ||
| 1652 | Flk_p29ING_FWD | GTGGACACACATGCACTCCT | JH | 6/1/2015 | 20 | 60 | O.lurida | p29ING4 Flanking | Q8C0D7 | |
| 1651 | Flk_p29ING_REV | AAGCAGACTCAGATTCAGGC | JH | 6/1/2015 | 20 | 58 | O.lurida | Q8C0D7 |
Using the Apex Red PCR Master Mix I created master mixes for each set of primers and ran them together.
Reagent Table
| Reaction_Components | Volume (ul) | Final Concentration |
| 2x Apex Red | 12.5 | 125 |
| Forward Primer (10uM) | 0.5 | 5 |
| Reverse Primer (10uM) | 0.5 | 5 |
| H20 | 10.5 | 105 |
| 1:2 cDNA | 1 | |
| 25 |
Using the final concentration I mixed each master mix going from largest to smallest volume.
I then pipetted 24 ul in each well followed by 1 ul of water or sample depending.
Then I ran the following PCR program:
| Temp | Time |
| 95 C | 5 min |
| 95 C | 30 sec |
| 55 C | 30 sec |
| 72 C | 30 sec |
| repeat steps 2-4 40 times | |
| 72 C | 3 min |
| 4 C | Hold |
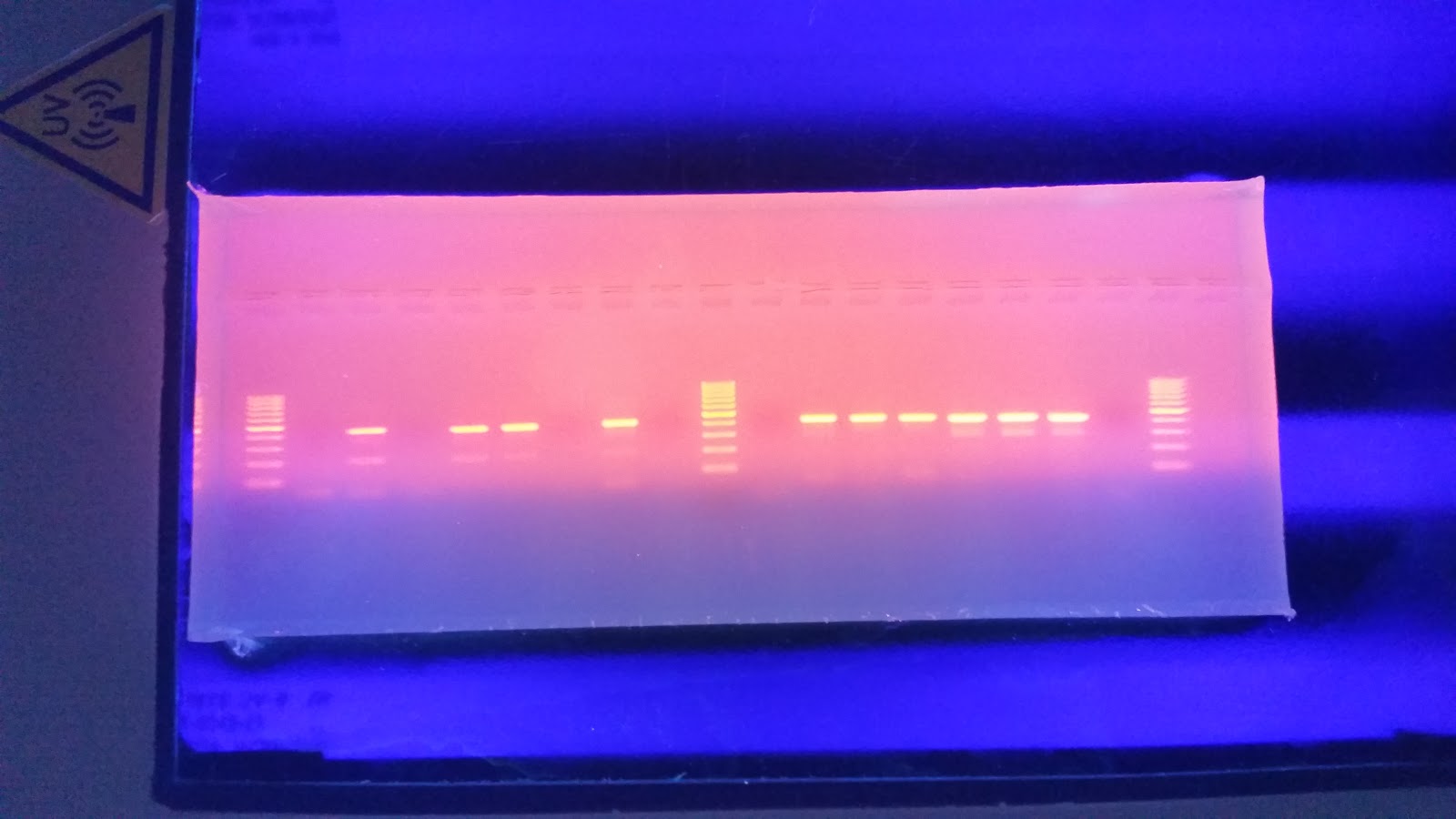
Once the PCR finished I ran the products on a 1.3% agarose gel
Gel Reagent Table
| Reagent | Volume |
| 1X Low TAE | 175-200 ml |
| Agarose | 2.3-2.6 g |
| EtBr | 17.5-20 ul |
- Add agarose to TAE.
- Microwave 1 minute stir
- Repeat until no particulate matter in solution
- Add EtBr while agarose still hot
- Gently pour in one corner of the gel cast until tray is full
I then ran the gels at 100 v for 35 minutes. I placed it on the transilluminator to view any bands that may have formed. The gels are below.
Gel 1 layout:
| PGRP-S | CARM | BMP2 | |||||||||||||||||
| Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 |
| BMP2 | HSPb11 | PGE/EP4 | |||||||||||||||||
| NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC |
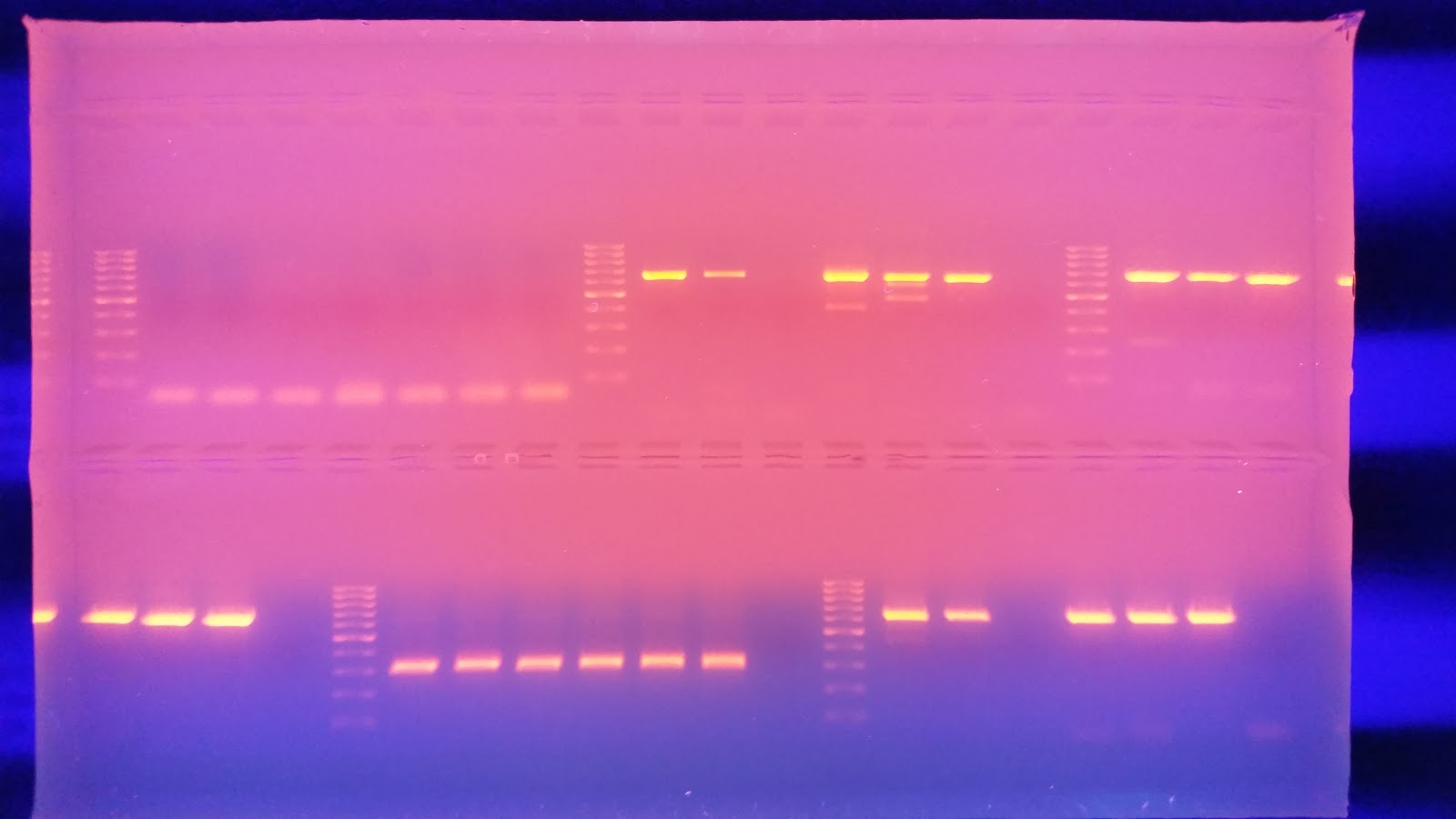
Gel 2 layout:
| GABABR | GRB2 | H3.3 | |||||||||||||||||
| Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 |
| H3.3 | H2A.V | H2A | |||||||||||||||||
| NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC |
| p291N4 | |||||||||||||||||||
| Ladder | NT1 | HT1 | ST1 | NC1 | HC1 | SC1 | NTC | Ladder | Empty | Empty | Empty | Empty | Empty | Empty | Empty | Empty | Empty | Empty | Empty |
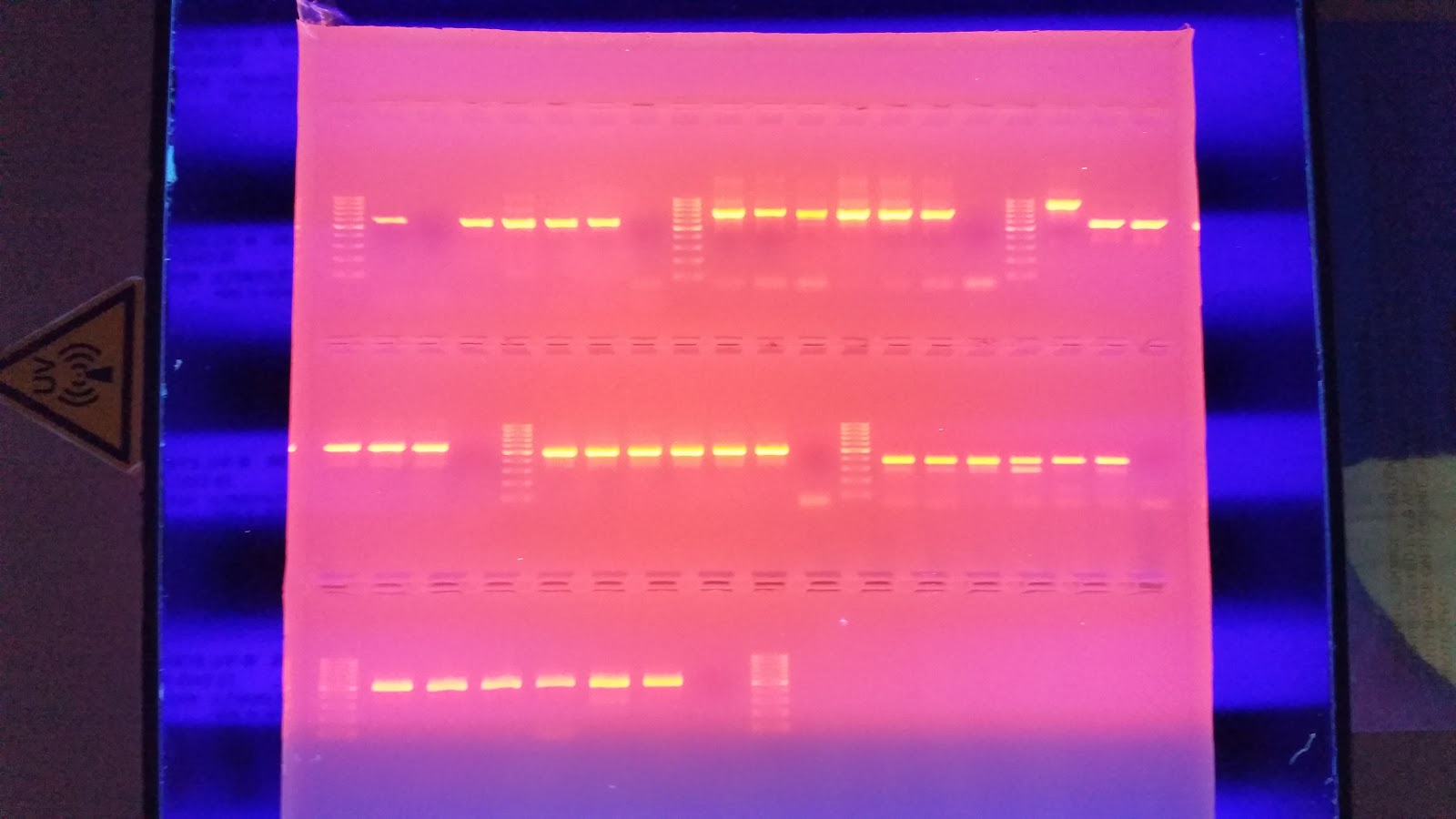
In Gel 1, the PGRP-S flanking primer failed completely. Also the Oyster Bay heat treated samples failed to amplify with the CARM and PGE/EP4 flanking primers. These did amplify previously so I'm not sure what it means.
In Gel 2, The Heat treated Dabob sample failed to amplify with the GABABR primer again not sure what it means. Interesting, the Fidalgo heat treated sample's PCR product in the H3.3 region has a much higher molecular weight. Steven suggested this could be an alternative splicing which would be interesting.
All bands except those in the failed PGRP primer were cut out and placed in either labelled 1.5 ml tubes or labelled Millipore Purification columns. The ones in the column were centrifuged for 10 minutes at 5000 rcf. The purified solutions and gel bits are stored in the 4 C fridge in 209. When we get more purification columns either I or Sam will spin down the remaining bands. Once the samples are isolated they will be sent off for sequencing. Hopefully the sequences will be the same in all populations so that the qPCR's can be trusted. That or they are so wildly different they expose significant differences in the genes between populations. Either way it should be interesting.