- 2023 66
- 2022 47
- 2021 48
- 2020 186
- 2019 165
- 2018 141
- 2017 80
- 2016 74
- 2015 153
- 2014 51
- 2013 13
- 2012 36
- 2011 85
- 2010 79
- 2009 235
- 2008 16
2023
qPCR Analysis - C.gigas Matt George Poly:IC Diploids
Ran a “quick and dirty” qPCR analysis for the qPCR’s I’d previously run:
Transcript Identification and Alignments - C.virginica RNAseq with NCBI Genome GCF_002022765.2 Using Hisat2 and Stringtie on Mox Again
qPCR - C.gigas PolyIC Diploid MgCl2
Using diploid MgCl2 control cDNA from 20230816, performed qPCR on the following primer sets selected by Steven (GitHub Issue), along with Cg_Actin as a potential normalizing gene:
Reverse Transcription - C.gigas PolyIC Diploid MgCl2
After quantifying diploid MgCl2 (Notebook) earlier today and proceeded to make cDNA. Reverse transcription was performed using oligo dT primers using M-MLV RT (Promega), per the manufacturer’s recommendations. Used 400ng of RNA in each reaction. I used 400ng (instead of the usual 100ng) to simplify pipetting for high concentration samples, without the need/time to dilute samples. All reactions were done on ice in 0.5uL PCR tubes.
RNA Quantification - C.gigas PolyIC Diploid MgCl2
Steven asked me to generate cDNA (GitHub Issue) from Crassostrea gigas (Pacific oyster) RNA, as part of Matt George’s polyIC project. I had previously quantified RNA from five diploid and five triploid samples (Notebook), but the qPCR results (Notebook) didn’t reveal much. Plus, the diploids and triploids weren’t the same treatment types (i.e. the diploids were poly:IC-injected, whereas the triploids were controls - so they weren’t really comparable). So, Steven asked that I find comparable set of samples so that we’d have either diploid controls or triploids which were poly:IC-injected. Looking in the freezer box for samples (Google sheet), I found four diploid MgCl2 control RNA samples. Before starting reverse transcription, I felt it would be best to quantify the RNA using the Qubit 3.0 using the Broad Range Assay. Steven and I discussed which samples to use earlier this week.
Daily Bits - August 2023
20230830
sRNA-seq Alignments - E5 Coral P.evermanni Using ShortStack on Mox
Steven asked that I run ShortStack on the E5 P.evermanni sRNA-seq (GitHub Issue) data we had. I used trimmed sRNA-seq reads from 20230620 (notebook entry) and the P.evermanni genome (Porites_evermanni_v1.fa) from https://www.genoscope.cns.fr/corals/genomes.html, along with the known, mature miRNAs FastA from https://www.mirbase.org/download/ (downloaded 20230628). The job was run on Mox.
Read Extractions - M.magister MEGAN FastA to FastQ Arthropoda and Unassigned Reads
After getting Arthropoda and Unassigned reads extracted to FastA format (notebook entry) using MEGAN6 Community Edition, the next step was to use the FastA files to extract reads in FastQ format. I used seqtk to do this. The process is documented in the Jupyter Notebook below.
DIAMOND BLASTx - C.virginica Genes on Mox
As part of annotating Ballgown transcripts for this project, I generated a genes FastA file on 20230726 (notebook entry). The next aspect of the annotation process was to BLASTx the genes to get SwissProt IDs, and, eventually gene ontology (GO) terms/IDs. To get SwissProt IDs, the genes were DIAMOND BLASTx’d against a Swiss/UniProt database of reviewed proteins (downloaded 20230727). The job was run on Mox.
qPCR - C.gigas polyIC
Using cDNA from 20230721, performed qPCR on the following primer sets selected by Steven (GitHub Issue), along with Cg_Actin as a potential normalizing gene:
Data Wrangling - C.virginica Genes Only FastA from Genes BED File Using gffread on Raven
I’ve been reviewing some of the CEABIGR (GitHub repo) data I’ve generated; specifically transcript count data/calcs. As part of that, I feel like we need/should annotate the transcripts to be able to make some more informed conclusions. Steven had previously performed annotations (Notebook), as well as Yaamini (GitHub Issue). However, there are shortcomings to both of the approaches each one utilized. Steven’s annotation relied only on coding sequences (CDS), while Yaamini’s utilized only mRNAs.
Read Extractions - M.magister MEGAN Arthropoda and Unassigned Reads to FastA
After converting DIAMOND BLASTx/MEGAN DAA files to RMA6 and looking at the taxonomic breakdown of each sample (notebook entry), it was decided we should extract reads from Arthropoda (and all taxonomies below) and all Unassigned reads from each sample. To do so, I used MEGAN6 Community Edition.
RNA Quantification - C.gigas PolyIC
Steven asked me to generate cDNA (GitHub Issue) from Crassostrea gigas (Pacific oyster) RNA, as part of Matt George’s polyIC project. Before starting reverse transcription, I felt it would be best to quantify the RNA using the Qubit 3.0 using the Broad Range Assay. Steven and I discussed which samples to use earlier this week.
Reverse Transcription - C.gigas PolyIC RNA
Quantified RNA earlier today and proceeded to make cDNA. Reverse transcription was performed using oligo dT primers using M-MLV RT (Promega), per the manufacturer’s recommendations. Used 400ng of RNA in each reaction. I used 400ng (instead of the usual 100ng) to simplify pipetting for high concentration samples, without the need/time to dilute samples. All reactions were done on ice in 0.5uL PCR tubes.
qPCR - C.gigas Ctenidia cDNA for Noah’s Heat-Mechanical Stress Project
Using cDNA from 20230713, performed qPCR on the following primer sets selected by Steven:
qPCR - C.gigas Ctenidia cDNA for Noah’s Heat-Mechanical Stress Project
Using cDNA from 20230713, performed qPCR on the following primer sets selected by Steven:
qPCR - C.gigas cDNA Primer Tests for Noah’s Heat-Mechanical Stress Project
After generating cDNA earlier today from Noah’s Crassostrea gigas (Pacific oyster) RNA (isolated 20230712), I ran qPCRs using the pooled cDNA sample to test out some primers that Steven pulled from the freezer.
Reverse Transcription - C.gigas RNA from Noah’s Heat-Mechanical Stress Project
After isolating ctenidia RNA on 20230712 and then quantifying the RNA earlier today, I needed to perform reverse transcription to have cDNA for subsequent qPCRs. Reverse transcription was performed using oligo dT primers using M-MLV RT (Promega), per the manufacturer’s recommendations. Used 100ng of RNA in each reaction. All reactions were done on ice in 0.5uL PCR tubes.
RNA Quantification - C.gigas RNA from Noah’s Heat-Mechanical Stress Project
I quantified Noah’s Crassostrea gigas (Pacific oyster) ctenidia RNA from yesterday (20230712) using the Roberts Lab Qubit 3 and the RNA High Sensitivity (HS) Assay. 1uL of sample was used for each measurement.
RNA Isolation - C.gigas Ctenidia from Noah’s Heat-Mechanical Stress Project
Steven asked that I help isolate RNA (GitHub Issue) for Noah’s summer project involving heat and mechanical stress (tumbling) of adult Crassostrea gigas (Pacific oyster). Here’s a brief overview of the process:
Daily Bits - July 2023
File Conversion - M.magister MEGANized DAA to RMA6
After the initial DIAMOND BLASTx and subsequent MEGANization(notebook) ran for 41 days, I attempted to open the extremely large files in the MEGAN6 GUI to get an overview of taxonomic breakdown. Due to the large file sizes (the smallest is 68GB!), the GUI consistently crashed. Also, each attempt took an hour or two before it would crash. Looking into this a bit more, I realized that I needed to convert the MEGANized DAA files to RMA6 format before attempting to import into the MEGAN6 GUI! Gah! The RMA6 files are significantly smaller (like “only” 2GB) and there should be ample memory to import them.
Data Received - Pacific Cod (G.macrocephalus) Sequencing Data from Novogene
Downloaded Pacific cod (_G.macrocephalus) sequencing data. I believe this is a NOAA project, and I don’t have any info about it. So, not much to report here.
sRNA-seq Alignments - E5 Coral A.pulchra P.meandrina Using ShortStack on Mox
Steven had asked that I align the coral E5 sRNA-seq reads using ShortStack (GitHub Issue). I previously trimmed the sRNA-seq reads to 35bp in length (notebook). Next up was to actually perform the alignments using ShortStack4. A.pulchra was aligned to the P.millepora genome, per this GitHub Issue. This was run on Mox.
Trimming and QC - E5 Coral sRNA-seq Data fro A.pulchra P.evermanni and P.meandrina Using FastQC flexbar and MultiQC on Mox
After downloading (notebook) and then reorganizing the E5 coral RNA-seq data from Azenta project 30-789513166 (notebook), and after testing some trimming options for sRNA-seq data (notebook), I opted to use the trimming software flexbar. I ran FastQC for initial quality checks, followed by trimming with flexbar, and then final QC with FastQC/MultiQC. This was performed on all three species in the data sets: A.pulchra, P.evermanni, and P.meandrina. All aspects were run on Mox. Final trimming length was set to 35bp.
ORF Identification - L.staminea De Novo Transcriptome Assembly v1.0 Using Transdecoder on Mox
After performing a de novo transcriptome assembly with L.staminea RNA-seq data, the Trinity assembly stats were quite a bit more “exaggerated” than normally expected. In an attempt to get a better sense of which contigs might be more useful candidates for downstream analysis, I decided to run the assembly through Transdecoder to identify open reading frames (ORFs). This was run on Mox.
Transcriptome Assembly - De Novo L.staminea Trimmed RNAseq Using Trinity on Mox
As part of this GitHub Issue to create a de novo transcriptome assembly from L.staminea RNA-seq data, I trimmed the reads earlier today. Next up is the actual do novo assembly. I performed this using Trinity on Mox.
Trimming - L.staminea RNA-seq Using FastQC fastp and MultiQC on Mox
Per this GitHub Issue, Steven asked me to perform a de novo transcriptome assembly on one set of paired FastQ from some little neck clam (L.staminea) RNA-seq. Prior to assembly, I needed to trim the FastQs.
Daily Bits - June 2023
20230605
Repeats Identification - P.meandrina Using RepeatMasker on Mox
Steven asked me to run RepeatMasker on the P.meandrina genome (GitHub Issue) for eventual use in developing a count matrix for transposable elements. The P.meandrina genome is from here:
Trimming and QC - E5 Coral sRNA-seq Trimming Parameter Tests and Comparisons
In preparation for FastQC and trimming of the E5 coral sRNA-seq data, I noticed that my “default” trimming settings didn’t produce the results I expected. Specifically, since these are sRNAs and the NEBNext® Multiplex Small RNA Library Prep Set for Illumina (PDF) protocol indicates that the sRNAs should be ~21 - 30bp, it seemed odd that I was still ending up with read lengths of 150bp. So, I tried a couple of quick trimming comparisons on just a single pair of sRNA FastQs to use as examples to get feeback on how trimming should proceed.
Data Wrangling - P.meandrina Genome GFF to GTF Using gffread
As part of getting P.meandrina genome info added to our Lab Handbook Genomic Resources page, I will index the P.meandrina genome file (Pocillopora_meandrina_HIv1.assembly.fasta) using HISAT2, but need a GTF file to also identify exon/intro splice sites. Since a GTF file is not available, but a GFF file is, I needed to convert the GFF to GTF. Used gffread to do this on my computer. Process is documented in Jupyter Notebook linked below.
FastQ QC and Trimming - E5 Coral RNA-seq Data for A.pulchra P.evermanni and P.meandrina Using FastQC fastp and MultiQC on Mox
After downloading and then reorganizing the E5 coral RNA-seq data from Azenta project 30-789513166, I ran FastQC for initial quality checks, followed by trimming with fastp, and then final QC with FastQC/MultiQC. This was performed on all three species in the data sets: A.pulchra, P.evermanni, and P.meandrina. All aspects were run on Mox.
Data Management - E5 Coral RNA-seq and sRNA-seq Reorganizing and Renaming
Downloaded the E5 coral sRNA-seq data from Azenta project 30-852430235 on 20230515 and the E5 coral RNA-seq data from Azenta project 30-789513166 on 20230516. The data required some reorganization, as the project included data from three different species (Acropora pulchra, Pocillopora meandrina, and Porites evermanni). Additionally, since the project was sequencing the same exact samples with both RNA-seq and sRNA-seq, the resulting FastQ files ended up being the same. This fact seemed like it could lead to potential downstream mistakes and/or difficulty tracking whether or not someone was actually using an RNA-seq or an sRNA-seq FastQ.
Data Received - Coral RNA-seq Data from Azenta Project 30-789513166
Small RNA-seq (sRNA-seq) data was made available from the coral E5 Azenta project 30-789513166. Sample sheet:
Data Received - Coral sRNA-seq Data from Azenta Project 30-852430235
Small RNA-seq (sRNA-seq) data was made available from the coral E5 Azenta project 30-852430235. Sample sheet is below.
lncRNA Expression - P.generosa lncRNA Expression Using StringTie
After identifying lncRNA in P.generosa, Steven asked that I generate an tissue-specific expression/count matrix (GitHub Issue). Looking through the documentation for StringTie, I decided that StringTie would work for this. The overall approach:
Daily Bits - May 2023
20230517
lncRNA Identification - P.generosa lncRNAs using CPC2 and bedtools
After trimming P.generosa RNA-seq reads on 20230426 and then aligning and annotating them to the Panopea-generosa-v1.0 genome on 20230426, I proceeded with the final step of lncRNA identification. To do this, I used Zach’s notebook entry on lncRNA identification for guidance. I utilized the annotated GTF generated by gffcompare during the alignment/annotation step on 20230426. I used ‘bedtools getfasta](https://bedtools.readthedocs.io/en/latest/content/tools/getfasta.html) and [CPC2` with an aribtrary 200bp minimum length to identify lncRNAs. All of this was done in a Jupyter Notebook (links below).
Containers - Apptainer Explorations
At some point, our HPC nodes on Mox will be retired. When that happens, we’ll likely purchase new nodes on the newest UW cluster, Klone. Additionally, the coenv nodes are no longer available on Mox. One was decommissioned and one was “migrated” to Klone. The primary issue at hand is that the base operating system for Klone appears to be very, very basic. I’d previously attempted to build/install some bioinformatics software on Klone, but could not due to a variety of missing libraries; these libraries are available by default on Mox… Part of this isn’t surprising, as UW IT has been making a concerted effort to get users to switch to containerization - specifically using Apptainer (formerly Singularity) containers.
Transcript Alignments - P.generosa RNA-seq Alignments for lncRNA Identification Using Hisat2 StingTie and gffcompare on Mox
This is a continuation of the process for identification of lncRNAs,. I aligned FastQs which were previously trimmed earlier today to our Panopea-generosa-v1.0 genome FastA using HISAT2. I used the HISAT2 genome index created on 20190723, which was created with options to identify exons and splice sites. The GFF used was from 20220323. StringTie was used to identify alternative transcripts, assign expression values, and create expression tables for use with ballgown. The job was run on Mox.
FastQ Trimming and QC - P.generosa RNA-seq Data from 20220323 on Mox
Addressing the update to this GitHub Issue regarding identifying Panopea generosa (Pacific geoduck) long non-coding RNAs (lncRNAs), I used the RNA-seq data from the Nextflow NF-Core RNAseq pipeline run on 20220323. Although that data was supposed to have been trimmed in the Nextflow NF-Core RNA-seq pipeline, the FastQC reports still show adapter contamination and some funky stuff happening at the 5’ end of the reads. So, I’ve opted to trim the “trimmed” files with fastp, using a hard 20bp trim at the 5’ end of all reads. FastQC and MultiQC were run before/after trimming. Job was run on Mox.
Data Wrangling - CEABIGR C.virginica Exon Expression Table
As part of the CEABIGR project (GitHub repo), Steven asked that I generate an exon expression table (GitHub Issue) where each row is a gene and the columns are the corresponding exons, filled with their expression value. For this, I planned on using the read count from the ballgown e_data.ctab files as an expression value.
Daily Bits - April 2023
20230403
Data Wrangling - Append Gene Ontology Aspect to P.generosa Primary Annotation File
Steven tasked me with updating our P.generosa genome annotation file (GitHub Issue) a while back and I finally managed to get it all figured out. Although I wanted to perform most of this using the GSEAbase package (PDF), as this package is geared towards storage/retrieval of gene set data, I eventually decided to abondon this approach due to the time it was taking and my lack of familiarity/understanding of how to manipulate objects in R. Despite that, GSEAbase was still utilized for its very simple use for identifying GOlims (IDs and Terms).
Sequencing Read Taxonomic Classification - M.magister RNA-seq Using DIAMOND BLASTx and MEGAN6 daa-meganizer on Mox
Running DIAMOND BLASTx, followed by MEGAN6 daa-meganizer for taxonomic classification of NOAA M.magister trimmed RNA-seq reads (provided by Giles Goetz on 20230301). This is primarily just for curiosity, per Steven’s GitHub Issue. This was run on Mox.
Daily Bits - March 2023
20230329
Data Received - Trimmed M.magister RNA-seq from NOAA
Sequencing Read Taxonomic Classification - P.verrucosa E5 RNA-seq Using DIAMOND BLASTx and MEGAN daa-meganizer on Mox
After some discussion with Steven at Science Hour last week regarding the handling of endosymbiont sequences in the E5 P.verrucosa RNA-seq data, Steven thought it would be interesting to run the RNA-seq reads through MEGAN6 just to see what the taxonomic breakdown looks like. We may or may not (probably not) separating reads based on taxonomy. In the meantime, we’ll still proceed with HISAT2 alignments to the respective genomes as a means to separate the endosymbiont reads from the P.verrucosa reads.
Data Wrangling - C.goreaui Genome GFF to GTF Using gffread
As part of getting these three coral species genome files (GitHub Issue) added to our Lab Handbook Genomic Resources page, I also need to get the coral endosymbiont sequence. After talking with Danielle Becker in Hollie Putnam’s Lab at Univ. of Rhode Island, she pointed me to the Cladocopium goreaui genome from Chen et. al, 2022 available here. Access to the genome requires agreeing to some licensing provisions (primarily the requirment to cite the publication whenever the genome is used), so I will not be providing any public links to the file. In order to index the Cladocopium goreaui genome file (Cladocopium_goreaui_genome_fa) using HISAT2 for downstream isoform analysis using StringTie and ballgown, I need a corresponding GTF to also identify exon/intro splice sites. Since a GTF file is not available, but a GFF file is, I needed to convert the GFF to GTF. Used gffread to do this on my computer. Process is documented in Jupyter Notebook linked below.
Transcript Identification and Alignments - P.verrucosa RNA-seq with Pver_genome_assembly_v1.0 Using HiSat2 and Stringtie on Mox
After getting the RNA-seq data trimmed, it was time to perform alignments and determine expression levels of transcripts/isoforms using with HISAT2 and StringTie, respectively. StringTie was set to output tables formatted for import into ballgown. After those two analyses were complete, I ran gffcompare, using the merged StringTie GTF and the input GFF3. I caught this in one of Danielle Becker’s scripts and thought it might be interesting. The analsyes were run on Mox.
FastQ Trimming and QC - P.verrucosa RNA-seq Data from Danielle Becker in Hollie Putnam Lab Using fastp FastQC and MultiQC on Mox
After receiving the P.verrucosa RNA-seq data from Danielle Becker (Hollie Putnam’s Lab, Univ. of Rhode Island), I noticed that the trimmed reads didn’t appear to actually be trimmed. There was still adapter contamination (solely in R2 reads - suggesting the detect_adapter_for_pe option had been omitted from the fastp command?), but the reads had an average read length of 150bp - except when looking at the adapter content report!!??.
Data Wrangling - Create P.verrucosa GCA_014529365.1 Karyotype File
Steven asked that I create a karyotype file (GitHub Issue) from the NCBI P.verrucosa genome (GCA_014529365.1) in the following format:
Data Received - P.verrucosa RNA-seq and WGBS Full Data from Danielle Becker
Worked with Danielle Becker, as part of the Coral E5 project, to transfer data related to her repo, from her HPC (Univ. of Rhode Island; Andromeda) to ours (Univ. of Washington; Mox) in order to eventually transfer to Gannet so that these files are publicly accessible to all members of the Coral E5 project.
Daily Bits - February 2023
20230223
Genome Indexing - P.verrucosa v1.0 Assembly with HiSat2 on Mox
Working on this Issue regarding adding coral genomes to our Handbook (GitHub) and needed to generate a HISAT2 index to add to The Roberts Lab Handbook Genomic Resources.
Genome Indexing - M.capitata HIv3 Assembly with HiSat2 on Mox
Working on this Issue regarding adding coral genomes to our Handbook (GitHub) and needed to generate a HISAT2 index to add to The Roberts Lab Handbook Genomic Resources.
Genome Indexing - P.acuta HIv2 Assembly with HiSat2 on Mox
Working on this Issue regarding adding coral genomes to our Handbook (GitHub) and needed to generate a HISAT2 index to add to The Roberts Lab Handbook Genomic Resources.
Data Wrangling - P.verrucosa Genome GFF to GTF Using gffread
As part of getting these three coral species genome files (GitHub Issue) added to our Lab Handbook Genomic Resources page, I will index the P.verrucosa genome file (Pver_genome_assembly_v1.0.fasta) using HISAT2, but need a GTF file to also identify exon/intro splice sites. Since a GTF file is not available, but a GFF file is, I needed to convert the GFF to GTF. Used gffread to do this on my computer. Process is documented in Jupyter Notebook linked below.
Data Wrangling - M.capitata Genome GFF to GTF Using gffread
As part of getting these three coral species genome files (GitHub Issue) added to our Lab Handbook Genomic Resources page, I will index the M.capitata genome file (Montipora_capitata_HIv3.assembly.fasta) using HISAT2, but need a GTF file to also identify exon/intro splice sites. Since a GTF file is not available, but a GFF file is, I needed to convert the GFF to GTF. Used gffread to do this on my computer. Process is documented in Jupyter Notebook linked below.
Data Wrangling - P.acuta Genome GFF to GTF Conversion Using gffread
As part of getting these three coral species genome files (GitHub Issue) added to our Lab Handbook Genomic Resources page, I will index the P.acuta genome file using HISAT2, but need a GTF file to also identify exon/intro splice sites. Since a GTF file is not available, but a GFF file is, I needed to convert the GFF to GTF. Used gffread to do this on my computer. Process is documented in Jupyter Notebook linked below.
Genome Indexing - P.verrucosa NCBI GCA_014529365.1 with HiSat2 on Mox
Working on this Issue regarding adding coral genomes to our Handbook (GitHub) and needed to generate a HISAT2 index to add to The Roberts Lab Handbook Genomic Resources.
Genome Indexing - M.capitata NCBI GCA_006542545.1 with HiSat2 on Mox
Working on this Issue regarding adding coral genomes to our Handbook (GitHub) and needed to generate a HISAT2 index to add to The Roberts Lab Handbook Genomic Resources.
SRA Data - Coral SRA BioProject PRJNA744403 Download and QC
Per this GitHub Issue, Steven wanted me to download all of the SRA data (RNA-seq and WGBS-seq) from NCBI BioProject PRJNA744403 and run QC on the data.
Monthly Goals - January 2023
- Plot CEABIGR coefficients of variation comparisons of mean gene methylation.
Daily Bits - January 2023
20230131
2022
BS-Seq Analysis - Nextflow EpiDiverse SNP Pipeline for Haws Hawaii C.gigas BAMs from Yaamini Base Config
Yaamini asked me to run the epidiverse/snp pipeline (GitHub Issue) on her Haws Crassostrea gigas (Pacific oyster) Hawaii bisuflite sequencing BAMs for SNP identification.
BS-Seq Analysis - Nextflow EpiDiverse SNP Pipeline for Haws Hawaii C.gigas BAMs from Yaamini
Yaamini asked me to run the epidiverse/snp pipeline (GitHub Issue) on her Haws Crassostrea gigas (Pacific oyster) Hawaii bisuflite sequencing BAMs for SNP identification.
Daily Bits - December 2022
20221227
Daily Bits - November 2022
20221121
Daily Bits - October 2022
20221031
Data Wrangling - C.virginica NCBI GCF_002022765.2 GFF to Gene and Pseudogene Combined BED File
Working on the CEABIGR project, I was preparing to make a gene expression file to use in CIRCOS (GitHub Issue) when I realized that the Ballgown gene expression file (CSV; GitHub) had more genes than the C.virginica genes BED file we were using. After some sleuthing, I discovered that the discrepancy was caused by the lack of pseudogenes in the genes BED file I was using. Although it might not really have any impact on things, I thought it would still be prudent to have a BED file that completely matched all of the genes in the Ballgown gene expression file. Plus, having the pseudogenes might be of longterm usefulness if we we ever decide to evalute the role of long non-coding RNAs (lncRNAs) in this project.
BSseq SNP Analysis - Nextflow EpiDiverse SNP Pipeline for C.virginica CEABIGR BSseq data
Steven asked that I identify SNPs from our C.virginica CEABIGR BSseq data (GitHub Issue). So, I ran sorted, deduplicated Bismark BAMs that Steven generated through the EpiDiverse/snp Nextflow pipeline. The job was run on Mox.
Data Wrangling - Identify C.virginica Genes with Different Predominant Isoforms for CEABIGR
During today’s discussion, Yaamini recommended we generate a list of genes with different predominant isoforms between females and males, while also adding a column with a binary indicator (e.g. 0 or 1) to mark those genes which were not different (0) or were different (1) between sexes. Steven had already generated files identifying predominant isoforms in each sex:
RNAseq Alignments - P.generosa Alignments and Alternative Transcript Identification Using Hisat2 and StringTie on Mox
As part of identifying long non-coding RNA (lncRNA) in Pacific geoduck(GitHub Issue), one of the first things that I wanted to do was to gather all of our geoduck RNAseq data and align it to our geoduck genome. In addition to the alignments, some of the examples I’ve been following have also utilized expression levels as one aspect of the lncRNA selection criteria, so I figured I’d get this info as well.
FastQ Trimming - Geoduck RNAseq Data Using fastp on Mox
Per this GitHub Issue, Steven asked me to identify long non-coding RNA (lncRNA) in geoduck. The first step is to aggregate all of our Panopea generosa (Pacific geoduck) RNAseq data and get it all trimmed. After that, align it to the genome, followed by Ballgown expression analysis, and then followed by a variety of selection criteria to parse out lncRNAs.
Daily Bits - September 2022
20220930
FastQ Trimming and QC - C.virginica Larval BS-seq Data from Lotterhos Lab and Part of CEABIGR Project Using fastp on Mox
We had some old Crassostrea virginica (Eastern oyster) larval/zygote BS-seq data from the Lotterhos Lab that’s part of the CEABiGR Workshop/Project (GitHub Repo) and Steven asked that I QC/trim it in this GitHub Issue.
Data Wrangling - Convert S.namaycush NCBI GFF to genes-only BED file for Use in Ballgown Analysis
In preparation for isoform identificaiton/quantification in S.namaycush RNAseq data, Ballgown will need a genes-only BED file. To generate, I used GFFutils to extract only genes from the NCBI GFF: GCF_016432855.1_SaNama_1.0_genomic.gff. All code was documented in the following Jupyter Notebook.
Splice Site Identification - S.namaycush Liver Parasitized and Non-Parasitized SRA RNAseq Using Hisat2-Stingtie with Genome GCF_016432855.1
After previously downloading/trimming/QCing S.namaycush SRA liver RNAseq data on 20220706, Steven asked that I run through Hisat2 for splice site identification (GitHub Issue).
qPCR - Repeat of Mussel Gill Heat Stress cDNA with Ferritin Primers
My previous qPCR on these cDNA using ferritin primers (SRIDs: 1808, 1809) resulted in no amplification. This was a bit surprising and makes me suspect that I screwed up somewhere (not adding primer(s)??), so I decided to repeat the qPCR. I made fresh working primer stocks and used 1uL of cDNA for each reaction. All reactions were run in duplicate on our CFX Connect thermalcycler (BioRad) with SsoFast EVAgreen Master Mix (BioRad). See my previous post linked above for qPCR master mix calcs.
Daily Bits - August 2022
20220831
qPCR - Dorothys Mussel cDNA from 20220726
Ran qPCRs on Dorothy’s mussel gill cDNA from 20220726 using the following primers:
RNA Isolation and Quantification - Dorothy Mussel Gill Samples
Isolated RNA from a subset of Dorothy’s mussel gill samples:
BS-seq and SNP Analysis - Nextflow EpiDiverse Pipelines Trials and Tribulations
Alrighty, this notebook entry is going to have a lot to unpack, as the process to get these pipelines running and then deal with the actual data we wanted to run them with was quite involved. However, the TL;DR of this all is this:
SRA Data - S.namaycush SRA BioProject PRJNA674328 Download and QC
Per this GitHub Issue, which I accidentally forgot about for three weeks (!), Steven wanted me to download the lake trout (Salvelinus namaycush) RNAseq data from NCBI BioProject PRJNA674328 and run QC on the data.
Daily Bits - July 2022
20220727
Daily Bits - June 2022
SRA Submission - Ostrea lurida MBD BS-seq from 20160203
Submitted our Ostrea lurida (Olympia oyster) MBD BS-seq data from 20160203 to the NCBI Sequence Read Archive (SRA).
Project Summary - C.virginica CEABiGR - Female vs. Male Gonad Exposed to OA
This will be a “dynamic” notebook entry, whereby I will update this post continually as I process new samples, analyze new data, etc for this project. The hope is to make it easier to find all the work I’ve done for this without having to search my notebook to find individual notebook entries.
Data Wrangling - Create Primary P.generosa Genome Annotation File
Steven asked me to create a canonical genome annotation file (GitHub Issue). I needed/wanted to create a file containing gene IDs, SwissProt (SP) IDs, gene names, gene descriptions, and gene ontology (GO) accessions. To do so, I utilized the NCBI BLAST and DIAMOND BLAST annotations generated by our GenSas P.generosa genome annotation. Per Steven’s suggestion, I used the best match (i.e. lowest e-value) for any given gene between the two files.
RNA Isolation - O.nerka Berdahl Brain Tissues
Server Maintenance - Fix Server Certificate Authentication Issues
We had been encounterings issues when linking to images in GitHub (e.g. notebooks, Issues/Discussions) hosted on our servers (primarily Gannet). Images always showed up as broken links and, with some work, we could see an error message related to server authentication. More recently, I also noticed that Jupyter Notebooks hosted on our servers could not be viewed in NB Viewer. Attempting to view a Jupyter Notebook hosted on one of our servers results in a 404 error, with a note regarding server certificate problems. Finally, the most annoying issue was encountered when running the shell programs wget to retrieve files from our servers. This program always threw an error regarding our server certificates. The only way to run wget without this error was to add the option --no-check-certificate (which, thankfully, was a suggestion by wget error message).
Nextflow - Trials and Tribulations of Installing and Using NF-Core RNAseq
INSTALLATION
Data Wrangling - P.generosa Genomic Feature FastA Creation
Steven wanted me to generate FastA files (GitHub Issue) for Panopea generosa (Pacific geoduck) coding sequences (CDS), genes, and mRNAs. One of the primary needs, though, was to have an ID that could be used for downstream table joining/mapping. I ended up using a combination of GFFutils and bedtools getfasta. I took advantage of being able to create a custom name column in BED files to generate the desired FastA description line having IDs that could identify, and map, CDS, genes, and mRNAs across FastAs and GFFs.
Differential Gene Expression - P.generosa DGE Between Tissues Using Nextlow NF-Core RNAseq Pipeline on Mox
Steven asked that I obtain relative expression values for various geoduck tissues (GitHub Issue). So, I decided to use this as an opportunity to try to use a Nextflow pipeline. There’s an RNAseq pipeline, NF-Core RNAseq which I decided to use. The pipeline appears to be ridiculously thorough (e.g. trims, removes gDNA/rRNA contamination, allows for multiple aligners to be used, quantifies/visualizes feature assignments by reads, performs differential gene expression analysis and visualization), all in one package. Sounds great, but I did have some initial problems getting things up and running. Overall, getting things set up to actually run took longer than the actual pipeline run! Oh well, it’s a learning process, so that’s not totally unexpected.
Data Analysis - C.virginica BSseq Unmapped Reads Using MEGAN6
After performing DIAMOND BLASTx and DAA “meganization” on 20220302, the next step was to import the DAA files into MEGAN6 for analyzing the resulting taxonomic assignments of the Crassostrea virginica (Eastern oyster) unmapped BSseq reads that Steven generated.
Data Analysis - C.virginica RNAseq Zymo ZR4059 Analyzed by ZymoResearch
After realizing that the Crassostrea virginica (Eastern oyster) RNAseq data had relatively low alignment rates (see this notebook entry from 20220224 for a bit more background), I contacted ZymoResearch to see if they had any insight on what might be happening. I suspected rRNA contamination. ZymoResearch was kind enough to run the RNAseq data through their pipeline and provided us. This notebook entry provides a brief overview and thoughts on the report.
Taxonomic Assignment - C.virginica BSseq Unmapped Reads Using DIAMOND BLASTx and MEGAN6 on Mox
After mapping bisulfite sequencing (BSseq) data to the Crassostrea virginica (Eastern oyster) genome, Steven noticed that there were a large number of unmapped reads. He asked that I attempt to taxonomically claissify the unmapped reads (GitHub Issue), with the idea that maybe these reads could provide additional data on an associated microbiome (GitHub Discussion).
Data Wrangling - P.generosa Genome GFF Conversion to GTF Using gffread
Steven asked in this GitHub Issue to convert our Panopea generosa (Pacific geoduck) genomic GFF to a GTF for use in the 10x Genomics Cell Ranger software. This conversion was performed using GffRead in a Jupyter Notebook.
Transcript Identification and Alignments - C.virginica RNAseq with NCBI Genome GCF_002022765.2 Using Hisat2 and Stringtie on Mox
After an additional round of trimming yesterday, I needed to identify alternative transcripts in the Crassostrea virginica (Eastern oyster) gonad RNAseq data we have. I previously used HISAT2 to index the NCBI Crassostrea virginica (Eastern oyster) genome and identify exon/splice sites on 20210720. Then, I used this genome index to run StringTie on Mox in order to map sequencing reads to the genome/alternative isoforms.
Trimming - Additional 20bp from C.virginica Gonad RNAseq with fastp on Mox
When I previously aligned trimmed RNAseq reads to the NCBI C.virginica genome (GCF_002022765.2) on 20210726, I specifically noted that alignment rates were consistently lower for males than females. However, I let that discrepancy distract me from a the larger issue: low alignment rates. Period! This should have thrown some red flags and it eventually did after Steven asked about overall alignment rate for an alignment of this data that I performed on 20220131 in preparation for genome-guided transcriptome assembly. The overall alignment rate (in which I actually used the trimmed reads from 20210714) was ~67.6%. Realizing this was a on the low side of what one would expect, it prompted me to look into things more and I came across a few things which led me to make the decision to redo the trimming:
Data Wrangling - C.virginica lncRNA Extractions from NCBI GCF_002022765.2 Using GffRead
Continuing to work on our Crassostrea virginica (Eastern oyster) project examining the effects of OA on female and male gonads (GitHub repo), Steven tasked me with parsing out long, non-coding RNAs (GitHub Issue). To do so, I relied on the NCBI genome and associated files/annotations. I used GffRead, GFFutils, and samtools. The process was documented in the followng Jupyter Notebook:
Transcriptome Assembly - Genome-guided C.virginica Adult Gonad OA RNAseq Using Trinity on Mox
As part of this project, Steven’s asked that I identify long, non-coding RNAs (lncRNAs) (GitHub Issue) in the Crassostrea virginica (Eastern oyster) adult OA gonad RNAseq data we have. The initial step for this is to assemble transcriptome. I generated the necessary BAM alignment on 20220131. Next was to actually get the transcriptome assembled. I followed the Trinity genome-guided procedure.
RNAseq Alignment - C.virginica Adult OA Gonad Data to GCF_002022765.2 Genome Using HISAT2 on Mox
As part of this project, Steven’s asked that I identify long, non-coding RNAs (lncRNAs) (GitHub Issue) in the Crassostrea virginica (Eastern oyster) adult OA gonad RNAseq data we have. The initial step for this is to assemble transcriptome. Since there is a published genome (NCBI RefSeq GCF_002022765.2C_virginica-3.0)](https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/) for [_Crassostrea virginica (Eastern oyster), I will perform a genome-guided assembly using Trinity. That process requires a sorted BAM file as input. In order to generate that file, I used HISAT2. I’ve already generated the necessary HISAT2 genome index files (as of 20210720), which also identified/incorporated splice sites and exons, which the HISAT2 alignment process requires to run.
Data Wrangling - C.virginica Gonad RNAseq Transcript Counts Per Gene Per Sample Using Ballgown
As we continue to work on the analysis of impacts of OA on Crassostrea virginica (Eastern oyster) gonads via DNA methylation and RNAseq (GitHub repo), we decided to compare the number of transcripts expressed per gene per sample (GitHub Issue). As it turns out, it was quite the challenge. Ultimately, I wasn’t able to solve it myself, and turned to StackOverflow for a solution. I should’ve just done this at the beginning, as I got a response (and solution) less than five minutes after posting! Regardless, the data wrangling progress (struggle?) was documented in the following GitHub Discussion:
Data Received - C.virginica OA Larvae DNA Methylation FastQs from Lotterhos Lab
In this GitHub Issue Steven asked that I download Crassostrea virginica (Eastern oyster) OA larval DNA methylation sequencing data from the Lotterhos Lab. The data is part of this project (private GitHub repo): epigeneticstoocean/2018_L18_OAExp_Cvirginica_DNAm. Alan Downey-Wall provided me with a GlobusConnect link to the data.
Project Summary - Matt George PSMFC Mytilus Byssus Project
This will be a “dynamic” notebook entry, whereby I will update this post continually as I process new samples, analyze new data, etc for this project. The hope is to make it easier to find all the work I’ve done for this without having to search my notebook to find individual notebook entries.
RNA Isolation - O.nerka Berdahl Brain Tissues
RNA Isolation - M.trossulus Gill
As part of a mussel project that Matt George has with the Pacific States Marine Fisheries Commission (PSMFC), I’m helping by continuing isolating RNA from a relatively large number of samples. The samples are listed/described in this GitHub Issue. Today, I isolated RNA from the following samples (the “F” indicates “foot”, “PG” indicates “phenol gland”, and “G” indicates “gill” tissues):
RNA Isolation - M.trossulus Gill and Phenol Gland
As part of a mussel project that Matt George has with the Pacific States Marine Fisheries Commission (PSMFC), I’m helping by continuing isolating RNA from a relatively large number of samples. The samples are listed/described in this GitHub Issue. Today, I isolated RNA from the following samples (the “F” indicates “foot”, “PG” indicates “phenol gland”, and “G” indicates “gill” tissues):
RNA Isolation - M.trossulus Gill and Phenol Gland
As part of a mussel project that Matt George has with the Pacific States Marine Fisheries Commission (PSMFC), I’m helping by continuing isolating RNA from a relatively large number of samples. The samples are listed/described in this GitHub Issue. Today, I isolated RNA from the following samples (the “F” indicates “foot”, “PG” indicates “phenol gland”, and “G” indicates “gill” tissues):
RNA Isolation - M.trossulus Gill and Phenol Gland
As part of a mussel project that Matt George has with the Pacific States Marine Fisheries Commission (PSMFC), I’m helping by continuing isolating RNA from a relatively large number of samples. The samples are listed/described in this GitHub Issue. Today, I isolated RNA from the following samples (the “F” indicates “foot”, “PG” indicates “phenol gland”, and “G” indicates “gill” tissues):
2021
RNA Isolation - M.trossulus Phenol Gland and Gill
As part of a mussel project that Matt George has with the Pacific States Marine Fisheries Commission (PSMFC), I’m helping by continuing isolating RNA from a relatively large number of samples. The samples are listed/described in this GitHub Issue. Today, I isolated RNA from the following samples (the “F” indicates “foot”, “PG” indicates “phenol gland”, and “G” indicates “gill” tissues):
RNA Isolation - M.trossulus Foot and Phenol Gland
As part of a mussel project that Matt George has with the Pacific States Marine Fisheries Commission (PSMFC), I’m helping by isolating RNA from a relatively large number of samples. The samples are listed/described in this GitHub Issue. Today, I isolated RNA from the following samples (the “F” indicates “foot” and the “PG” indicates “phenol gland” tissues):
Project Summary - O.nerka Berdahl Samples
This will be a “dynamic” notebook entry, whereby I will update this post continually as I process new samples, analyze new data, etc for this project. The hope is to make it easier to find all the work I’ve done for this without having to search my notebook to find individual notebook entries.
RNA Isolation - O.nerka Berdahl Brain Tissues
RNA Isolation - O.nerka Berdahl Tissues
Finally got around to tackling this GitHub issue regarding isolating RNA from some Oncorhynchus nerka (sockeye salmon) tissues we have from Andrew Berdahl’s lab (a UW SAFS professor) to use for RNAseq and/or qPCR. We have blood, brain, gonad, and liver samples from individual salmon from two different groups: territorial and social individuals. We’ve decided to isolate RNA from brain, gonads, and liver from two individuals within each group. All samples are preserved in RNAlater and stored @ -80oC.
Data Wrangling - C.virginica NCBI GCF_002022765.2 GFF to Gene BED File
When working to identify differentially expressed transcripts (DETs) and genes (DEGs) for our Crassostrea virginica (Eastern oyster) RNAseq/DNA methylation comparison of changes across sex and ocean acidification conditions (https://github.com/epigeneticstoocean/2018_L18-adult-methylation), I realized that the DEG tables I was generating had excessive gene counts due to the fact that the analysis (and, in turn, the genome coordinates), were tied to transcripts. Thus, genes were counted multiple times due to the existence of multiple transcripts for a given gene, and the analysis didn’t list gene coordinate data - only transcript coordinates.
Differential Transcript Expression - C.virginica Gonad RNAseq Using Ballgown
In preparation for differential transcript analysis, I previously ran our RNAseq data through StringTie on 20210726 to identify and quantify transcripts. Identification of differentially expressed transcripts (DETs) and genes (DEGs) will be performed using ballgown. This notebook entry will be different than most others, as this notebook entry will simply serve as a “landing page” to access/review the analysis; as the analysis will evolve over time and won’t exist as a single computing job with a definitive endpoint.
SNP Characterization - C.bairdi v3.1 Transcriptome Assembly and Day2 DEG Pooled Samples RNAseq Data
I previously identified variants across the four Day 2 DEG pooled RNAseq samples (380822, 380823, 380824, 380825) on 20210909 within the cbai_transcriptome_v3.1 transcriptome assembly. Now, I needed to actually do some analysis on the SNPs for the revisions to the Tanner crab gene expression paper (GitHub Repo).
SNP Identification - C.bairdi Day 2 DEG Pooled Samples Using bcftools on Mox
RNAseq Alignments - C.bairdi Day 2 Infected-Uninfected Temperature Increase-Decrease RNAseq to cbai_transcriptome_v3.1.fasta with Hisat2 on Mox
Ealier today, I created the necessary Hisat2 index files for cbai_transcriptome_v3.1. Next, I needed to actually get the alignments run. The alignments were performed using HISAT2 on Mox using the following set of trimmed FastQ files from 2020414:
Assembly Indexing - C.bairdi Transcriptome cbai_transcriptome_v3.1.fasta with Hisat2 on Mox
We recently received reviews back for the Tanner crab paper submission (“Characterization of the gene repertoire and environmentally driven expression patterns in Tanner crab (Chionoecetes bairdi)”) and one of the reviewers requested a more in-depth analysis. As part of addressing this, we’ve decided to identify SNPs withing the _Chionoecetes bairdi (Tanner crab) transcriptome used in the paper (cbai_transcriptome_v3.1). Since the process involves aligning sequencing reads to the transcriptome, the first thing that needed to be done was to generate index files for the aligner (HISAT2, in this particular case), so I ran HISAT2 on Mox.
Computer Servicing - APC SUA2200RM2U UPS Battery Replacement
Replaced the battery pack in the top APC SUA2200RM2U UPS in our computing “cluster” cabinet.
Computer Management - Disable Sleep and Hibernation on Raven
We’ve been having an issue with our computer Raven where it would become inaccessible after some time after a reboot. Attempts to remote in would just indicate no route to host or something like that. We realized it seemed like this was caused by a power saving setting, but changing the sleep setting in the Ubuntu GUI menu didn’t fix the issue. It also seemd like the sleep/hibernate issue was only a problem after the computer had been rebooted and no one had logged in yet…
Uninterruptible Power Supply - Battery Replacement for APS BR1000G
Replace battery in uninterruptible power supply APS BR1000G for Owl in FTR 230.
Transcript Identification and Quantification - C.virginia RNAseq With NCBI Genome GCF_002022765.2 Using StringTie on Mox
After having run HISAT2 to index and identify exons and splice sites in the NCBI Crassostrea virginica (Eastern oyster) genome (GCF_002022765.2) on 20210720, the next step was to identify and quantify transcripts from the RNAseq data using StringTie.
Genome Annotations - Splice Site and Exon Extractions for C.virginica GCF_002022765.2 Genome Using Hisat2 on Mox
Previously performed quality trimming on the Crassostrea virginica (Eastern oyster) gonad/sperm RNAseq data on 20210714. Next, I needed to identify exons and splice sites, as well as generate a genome index using HISAT2 to be used with StringTie downstream to identify potential alternative transcripts. This utilized the following NCBI genome files:
Trimming - C.virginica Gonad RNAseq with FastP on Mox
Needed to trim the Crassostrea virginica (Eastern oyster) gonad RNAseq data we received on 20210528.
Summary - Geoduck RNAseq Data
Per this GitHub Issue, I’ve compiled a summary table, with links, of all of our Panopea generosa (Pacific geoduck) RNAseq data as it exists in NCBI. This will be a “dynamic” notebook entry, whereby I will update this post continually as we acquire new data and/or change the information we’d like to have here.
Genome Analysis - Identification of Potential Contaminating Sequences in Panopea-generosa-v1.0 Assembly Using BlobToolKit on Mox
As part of our Panopea generosa (Pacific geoduck) genome sequencing efforts, Steven came across a tool designed to help identify if there are any contaminating sequences in your assembly. The software is BlobToolKit. The software is actually a complex pipeline of separate tools ([minimap2])https://github.com/lh3/minimap2, BLAST, DIAMOND BLAST, and BUSCO) which aligns sequencing reads and assigns taxonomy to the reads, as well as marking regions of the assembly with various taxonomic assignments.
FastQC-MultiQC - Yaamini’s C.virginica RNAseq and WGBS from ZymoResearch on Mox
Finally got around to running FastQC on Yaamini’s RNAseq and WGBS sequencing data recieved on 20210528.
Data Wrangling - S.salar Gene Annotations from NCBI RefSeq GCF_000233375.1_ICSASG_v2_genomic.gff for Shelly
Shelly posted a GitHub Issue asking if I could create a file of S.salar genes with their UniProt annotations (e.g. gene name, UniProt accession, GO terms).
Data Received - Yaamini’s C.virginica WGBS and RNAseq Data from ZymoResearch
Yaamini received her sequencing data from ZymoResearch; both whole genome bisfulfite sequencing (WGBS) and RNAseq. I was tasked with downloading the data and running QC.
Assembly Comparisons - Ostrea lurida Non-scaffold Genome Assembly Comparisons Using Quast on Swoose
After generating a new Ostrea lurida genome assembly (v090) on 20210520, I decided to compare with our previous genome assemblies. Here, I compared v090 with all of our previous scaffolded assemblies using Quast. Here is a table (GitHub) which describes all of our existing assemblies (i.e. assembly name, assembly process, etc):
Assembly Assessment - Olurida_v090 Using Quast on Swoose
After running a new Ostrea lurida assembly yesterday (Olurida_v090), I evaluated Olurida_v090 using Quast to produce some stats. This was run on my local computer with the following command:
Genome Assembly - Olurida_v090 with BGI Illumina and PacBio Hybrid Using Wengan on Mox
I was recently tasked with adding annotations for our Ostrea lurida genome assembly to NCBI. As it turns out, adding just annotation files can’t be done since the genome was initially submitted to ENA. Additionally, updating the existing ENA submission with annotations is not possible, as it requires a revocation of the existing genome assembly; requiring a brand new submission. With that being the case, I figured I’d just make a new genome submission with the annotations to NCBI. Unfortunately, there were a number of issues with our genome assembly that were going to require a fair amount of work to resolve. The primary concern was that most of the sequences are considered “low quality” by NCBI (too many and too long stretches of Ns in the sequences). Revising the assembly to make it compatible with the NCBI requirements was going to be too much, so that was abandoned.
Trimming - O.lurida BGI FastQs with FastP on Mox
After attempting to submit our Ostrea lurida (Olympia oyster) genome assembly annotations (via GFF) to NCBI, the submission process also highlighted some short comings of the Olurida_v081 assembly. When getting ready to submit the genome annotations to NCBI, I was required to calculate the genome coverage we had. NCBI suggested to calculate this simply by counting the number of bases sequenced and divide it by the genome size. Doing this resulted in an estimated coverage of ~55X coverage, yet we have significant stretches of Ns throughout the assembly. I understand why this is still technically possible, but it’s just sticking in my craw. So, I’ve decided to set up a quick assembly to see what I can come up with. Of note, the canonical assembly we’ve been using relied on the scaffolded assembly provided by BGI; we never attempted our own assembly from the raw data.
Genome Submission - Validation of Olurida_v081.fa and Annotated GFFs Prior to Submission to NCBI
Per this GitHub Issue, Steven has asked to get our Ostrea lurida (Olympia oyster) genome assembly (Olurida_v081.fa) submitted to NCBI with annotations. The first step in the submission process is to use the NCBI table2asn_GFF software to validate the FastA assembly, as well as the GFF annotations file. Once the software has been run, it will point out any errors which need to be corrected prior to submission.
RepeatMasker - C.gigas Rosling NCBI Genome GCA_902806645.1 on Mox
Decided to tackle this GitHub Issue about creating a transposable elements IGV track with the new Roslin C.gigas genome, since it had been sitting for a while and I have code sitting around that’s ready to roll for this type of thing.
Singularity - RStudio Server Container on Mox
Aidan recently needed to use R on a machine with more memory. Additionally, it would be ideal if he could use RStudio. So, I managed to figure out how to set up a Singularity container running rocker/rstudio.
Read Mapping - 10x-Genomics Trimmed FastQ Mapped to P.generosa v1.0 Assembly Using Minimap2 for BlobToolKit on Mox
To continue towards getting our Panopea generosa (Pacific geoduck) genome assembly (v1.0) analyzed with BlobToolKit, per this GitHub Issue, I’ve decided to run each aspect of the pipeline manually, as I continue to have issues utilizing the automatic pipeline. As such, I’ve run minimap2 according to the BlobToolKit “Getting Started” guide on Mox. This will map the trimmed 10x-Genomics reads from 20210401 to the Panopea-generosa-v1.0.fa assembly (FastA; 914MB).
Genome Annotation - P.generosa v1.0 Assembly Using DIAMOND BLASTx for BlobToolKit on Mox
To continue towards getting our Panopea generosa (Pacific geoduck) genome assembly (v1.0) analyzed with BlobToolKit, per this GitHub Issue, I’ve decided to run each aspect of the pipeline manually, as I continue to have issues utilizing the automatic pipeline. As such, I’ve run DIAMOND BLASTx according to the BlobToolKit “Getting Started” guide on Mox.
Genome Annotation - P.generosa v1.0 Assembly Using BLASTn for BlobToolKit on Mox
To continue towards getting our Panopea generosa (Pacific geoduck) genome assembly (v1.0) analyzed with BlobToolKit, per this GitHub Issue, I’ve decided to run each aspect of the pipeline manually, as I continue to have issues utilizing the automatic pipeline. As such, I’ve run BLASTn according to the BlobToolKit “Getting Started” guide on Mox.
Trimming P.generosa 10x Genomics HiC FastQs with fastp on Mox
Steven asked me to try running Blob Tool Kit to identify potential contaminating sequence in our Panopea generosa (Pacific geoduck) genome assembly (v1.0). In preparation for running Blob Tool Kit, I needed to trim the 10x Genomics FastQ data used by Phase Genomics. Files were trimmed using fastp on Mox.
Transcriptome Annotation - Trinotate on C.bairdi Transcriptome v4.0 on Mox
Continued annotation of cbai_transcriptome_v4.0.fasta [Trinity de novo assembly from 20210317(https://robertslab.github.io/sams-notebook/2021/03/17/Transcriptome-Assembly-C.bairdi-Transcriptome-v4.0-Using-Trinity-on-Mox.html)] using Trinotate on Mox. This will provide a thorough annotation, including genoe ontology (GO) term assignments to each contig.
Transcriptome Annotation - DIAMOND BLASTx on C.bairdi Transcriptome v4.0 on Mox
Continued annotation of cbai_transcriptome_v4.0.fasta [Trinity de novo assembly from 20210317(https://robertslab.github.io/sams-notebook/2021/03/17/Transcriptome-Assembly-C.bairdi-Transcriptome-v4.0-Using-Trinity-on-Mox.html)] using DIAMOND BLASTx on Mox. This will be used as a component of Trinotate annotation downstream.
TransDecoder - C.bairdi Transcriptome v4.0 on Mox
Began annotation of cbai_transcriptome_v4.0.fasta Trinity de novo assembly from 20210317] using TransDecoder on Mox. This will be used as a component of Trinotate annotation downstream.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi Transcriptome v4.0 on Mox
I previously created a C.bairdi de novo transcriptome assembly v4.0 with Trinity from all our C.bairdi RNAseq reads which had BLASTx matches to the C.opilio genome and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Assembly - C.bairdi Transcriptome v4.0 Using Trinity on Mox
Continuing to addressing this GitHub issue, to generate an additional C.bairdi transcriptome, I finally got to the point of actually running the assembly using Trinity using the extracted reads from 20210316. Those reads were identified via BLASTx agianst the C.opilio genome proteins on 20210312. Trinty was run on Mox.
Read Extractions - C.bairdi RNAseq Reads from C.opilio BLASTx Matches with seqkit on Mox
As part of addressing this GitHub issue, to generate an additional C.bairdi transcriptome, I needed to extract the reads ID’ed via BLASTX against the C.opilio genome on 20210312. Read extractions were performed using SeqKit on Mox.
DIAMOND BLASTx - C.bairdi RNAseq vs C.opilio Genome Proteins on Mox
We want to generate an additional Tanner crab (Chionoecetes bairdi) transcriptome, per this GitHub issue, to generate an additional C.bairdi transcriptome. This has come about due to the release of the genome of a very closely related crab species, Chionoecetes opilio (Snow crab).
Transcriptome Annotation - Trinotate Hematodinium v1.7 on Mox
Transcriptome Annotation - Trinotate Hematodinium v1.6 on Mox
Transcriptome Assembly - Hematodinium Transcriptomes v1.6 and v1.7 with Trinity on Mox
I’d previously assembled hemat_transcriptome_v1.0.fasta on 20200122, hemat_transcriptome_v1.5.fasta on 20200408, extracted hemat_transcriptome_v2.1.fasta from an existing FastA on 20200605, as well as extracted hemat_transcriptome_v3.1.fasta on 20200605.
Data Wrangling - Gene ID Extraction from P.generosa Genome GFF Using Methylation Machinery Gene IDs
Per this GitHub issue, Steven provided a list of methylation-related gene names and wanted to extract the corresponding Panopea generosa ([Pacific geoduck (Panopea generosa)](http://en.wikipedia.org/wiki/Geoduck)) gene ID from our P.generosa genome, along with corresponding BLAST e-values.
Data Wrangling - Gene ID Extraction from P.generosa Genome GFF Using Methylation Machinery List
Per this GitHub Issue Steven asked that I take a list of gene names associated with DNA methylation and see if I could extract a list of Panopea generosa (Panopea generosa) gene IDs and corresponding BLAST e-values for each from our P.generosa genome annotation (see Genomic Resources wiki for more info).
SRA Submission - A.elegantissima ONT Fast5 from Jay Dimond
At the beginning of the month, Jay sent us his A.elegantissima ONT Fast5 data in order to help him get it submitted to NCBI Sequencing Read Archive (SRA).
Data Received - Anthopleura elegantissima - aggregating anenome - NanoPore Genome Sequence from Jay Dimond
Jay asked me to help get his A.elegantissima (aggregating anenome) NanoPore gDNA sequencing data submitted to NCBI Sequencing Read Archive (SRA). He sent a hard drive (HDD) with all the NanoPore sequencing Fast5 files. The HDD was received on 2/2/2021. Here’re are details provided in the reamde file in the Ae_ONT directory.
Samples Submitted - M.magister MBD-BSseq Libraries to Univ. of Oregon GC3F
Submitted the M.magister MBD-BSseq libraries created 20201124 using the 4nM aliquots created for the MiSeq test run on 20201202 to the Univ. of Oregon GC3F sequencing core.
2020
Transcriptome Comparisons - C.bairdi Transcriptomes Evaluations with DETONATE rsem-eval on Mox
UPDATE: I’ll lead in with the fact that this failed with an error message that I can’t figure out. This will save the reader some time. I’ve posted the problem as an Issue on the DETONATE GitHub repo, however it’s clear that this software is no longer maintained, as the repo hasn’t been updated in >3yrs; even lacking responses to Issues that are that old.
Alignments - C.bairdi RNAseq Transcriptome Alignments Using Bowtie2 on Mox
I had previously attempted to compare all of our C.bairdi transcriptome assemblies using DETONATE on 20200601, but, due to hitting time limits on Mox, failed to successfully get the analysis to complete. I realized that the limiting factor was performing FastQ alignments, so I decided to run this step independently to see if I could at least get that step resolved. DETONATE (rsem-eval) will accept BAM files as input, so I’m hoping I can power through this alignment step and then provided DETONATE (rsem-eval) with the BAM files.
Samples Received - Cockle Clam Gonad H and E Slides
Today we received the H & E-stained slides from the cockle clam gonad tissue blocks/cassettes we submitted on 20201201. Slides were added to Slide Case #5 - Rows 13 - 37 (Google Sheet).
FastQC-MultiQC - M.magister MBD-BSseq Pool Test MiSeq Run on Mox
Earlier today we received the M.magister (C.magister; Dungeness crab) MiSeq data from Mac.
Data Received - M.magister MBD-BSseq Pool Test MiSeq Run
After creating _M.magister (C.magister; Dungeness crab) MBD-BSseq libraries (on 20201124), I gave the pooled set of samples to Mac for a test sequencing run on the MiSeq on 20201202.
Alignment - C.gigas RNAseq to GCF_000297895.1_oyster_v9 Genome Using STAR on Mox
Mac was getting some weird results when mapping some single cell RNAseq data to the C.gigas mitochondrial (mt) genome that she had, so she asked for some help mapping other C.gigas RNAseq data (GitHub Issue) to the C.gigas mt genome to see if someone else would get similar results.
SRA Submission - Haws Lab C.gigas Ploidy pH WGBS
Trimming - Haws Lab C.gigas Ploidy pH WGBS 10bp 5 and 3 Prime Ends Using fastp and MultiQC on Mox
Making the assumption that the 24 C.gigas ploidy pH WGBS data we receved 20201205 will be analyzed using Bismark, I decided to go ahead and trim the files according to Bismark guidelines for libraries made with the ZymoResearch Pico MethylSeq Kit.
FastQC-MultiQc - C.gigas Ploidy pH WGBS Raw Sequence Data from Haws Lab on Mox
Yesterday (20201205), we received the whole genome bisulfite sequencing (WGBS) data back from ZymoResearch from the 24 C.gigas diploid/triploid subjected to two different pH treatments (received from the Haws’ Lab on 20200820 that we submitted to ZymoResearch on 20200824. As part of our standard sequencing data receipt pipeline, I needed to generate FastQC files for each sample.
Data Received - C.gigas Diploid-Triploid pH Treatments Ctenidia WGBS from ZymoResearch
Today we received the whole genome bisulfite sequencing (WGBS) from the 24 C.gigas diploid-triploid samples subjected to different pH that were submitted 20200824. The lengthy turnaround time was due to a bad lot of reagents, which forced them Zymo to find a different manufacturer in order to generate libraries.
Trimming - Ronits C.gigas Ploidy WGBS 10bp 5 and 3 Prime Ends Using fastp and MultiQC on Mox
Steven asked me to trim (GitHub Issue) Ronit’s WGBS sequencing data we received on 20201110, according to Bismark guidelines for libraries made with the ZymoResearch Pico MethylSeq Kit.
Sample Submission - M.magister MBD BSseq Libraries for MiSeq at NOAA
Earlier today I quantified the libraries with the Qubit in preparation for sample pooling and sequencing. Before performing a full sequencing run, Mac wanted to select a subset of the libraries based on the experimental treatments to have an equal representation of samples. She also wanted to do a quick run on the MiSeq at NOAA to evaluate how well libraries map and to make sure libraries appear to be sequencing at relatively equal levels.
Library Quantification - M.magister MBD BSseq Libraries with Qubit
After reviewing the Bionalyzer assays for the MBD BSseq libraries Mac indicated she’d like to have the libraries quantified using the Qubit.
Samples Submitted - Cockle Clam Gonad Histology Cassettes for H and E
Per this GitHub Issue, Steven asked that I submit a set of fixed cockle clam gonad tissues (currently stored in histology cassettes in 70% EtOH) for Hematoxylin and eosin stain (H&E). I submitted the following samples to the UW Pathology Research Services Laboratory (UW PRSL):
Trimming - Ronits C.gigas Ploidy WGBS Using fastp and MultiQC on Mox
Steven asked me to trim (GitHub Issue) Ronit’s WGBS sequencing data we received on 20201110, according to Bismark guidelines for libraries made with the ZymoResearch Pico MethylSeq Kit.
Bioanalyzer - M.magister MBD BSseq Libraries
MBD BSseq library construction was completed yesterday (20201124). Next, I needed to evaluate the libraries using the Roberts Lab Bioanalyzer 2100 (Agilent) to assess library sizes, yields, and qualities (i.e. primer dimers).
MBD BSseq Library Prep - M.magister MBD-selected DNA Using Pico Methyl-Seq Kit
After finishing the final set of eight MBD selections on 20201103, I’m finally ready to make the BSseq libraries using the Pico Methyl-Seq Library Prep Kit (ZymoResearch) (PDF). I followed the manufacturer’s protocols with the following notes/changes (organized by each section in the protocol):
RNA Isolation and Quantification - P.generosa Hemocytes from Shelly
Shelly asked me to isolate RNA from some P.generosa hemocytes (GitHub Issue) that she had.
SRA Submission - Ronits C.gigas Ploidy WGBS
FastQC-MultiQc - C.gigas Ploidy WGBS Raw Sequence Data from Ronits Project on Mox
Transcriptome Assessment - Crustacean Transcripome Completeness Evaluation Using BUSCO on Mox
Grace was recently working on writing up a manuscript which did a basic comparison of our C.bairdi transcriptome (cbai_transcriptome_v3.1) (see the Genomic Resources wiki for more deets) to two other species’ transcriptome assemblies. We wanted BUSCO evaluations as part of this comparison, but the two other species did not have BUSCO scores in their respective publications. As such, I decided to generate them myself, as BUSCO runs very quickly. The job was run on Mox.
Data Received - C.gigas Ploidy WGBS from Ronits Project via ZymoResearch
We received the data from our whole genome bisulfite sequencing (WGBS) submission to ZymoResearch on 2020820 for Ronit’s C.gigas diploid/triploid dessication/heat stress project.
Data Wrangling - MultiQC on S.salar RNAseq from fastp and HISAT2 on Mox
In Shelly’s GitHub Issue for this S.salar project, she also requested a MultiQC report for the trimming (completed on 20201029) and the genome alignments (completed on 20201103).
Hard Drive Upgrade - Gannet Synology Server
Completed upgrading the 12 x 8TB HDDs in our server, Gannet (Synology RS3618XS), to 12 x 16TB HDDs. The process was simple, but the repair process took ~20hrs for each new drive. So, the entire process required 12 separate days of pulling out one old HDD, replacing with a new HDD, and initiating the repair process in the Synology web interface.
RNAseq Alignments - S.salar HISAT2 BAMs to GCF_000233375.1_ICSASG_v2_genomic.gtf Transcriptome Using StringTie on Mox
This is a continuation of addressing Shelly Trigg’s (regarding some Salmo salar RNAseq data) request (GitHub Issue) to trim (completed 20201029), perform genome alignment (completed on 20201103), and transcriptome alignment.
RNAseq Alignments - Trimmed S.salar RNAseq to GCF_000233375.1_ICSASG_v2_genomic.fa Using Hisat2 on Mox
This is a continuation of addressing Shelly Trigg’s (regarding some Salmo salar RNAseq data) request (GitHub Issue) to trim (completed 20201029), perform genome alignment, and transcriptome alignment.
MBD Selection - M.magister Sheared Gill gDNA 16 of 24 Samples Set 3 of 3
Click here for notebook on the first eight samples processed. Click here for the second set of eight samples processed. M.magister (Dungeness crab) gill gDNA provided by Mackenzie Gavery was previously sheared on 20201026 and three samples were subjected to additional rounds of shearing on 20201027, in preparation for methyl bidning domain (MBD) selection using the MethylMiner Kit (Invitrogen).
MBD Selection - M.magister Sheared Gill gDNA 8 of 24 Samples Set 2 of 3
Click here for notebook on the first eight samples processed. M.magister (Dungeness crab) gill gDNA provided by Mackenzie Gavery was previously sheared on 20201026 and three samples were subjected to additional rounds of shearing on 20201027, in preparation for methyl bidning domain (MBD) selection using the MethylMiner Kit (Invitrogen).
Trimming - Shelly S.salar RNAseq Using fastp and MultiQC on Mox
Shelly asked that I trim, align to a genome, and perform transcriptome alignment counts in this GitHub issue with some Salmo salar RNAseq data she had and, using a subset of the NCBI Salmo salar RefSeq genome, GCF_000233375.1. She created a subset of this genome using only sequences designated as “chromosomes.” A link to the FastA (and a link to her notebook on creating this file) are in that GitHub issue link above. The transcriptome she has provided has not been subsetted in a similar fashion; maybe I’ll do that prior to alignment.
MBD Selection - M.magister Sheared Gill gDNA 8 of 24 Samples Set 1 of 3
DNA Shearing - M.magister gDNA Additional Shearing CH05-01_21 CH07-11 and Bioanalyzer
After shearing all of the M.magister gill gDNA on 20201026, there were still three samples that still had average fragment lengths that were a bit longer than desired (~750bp, but want ~250 - 550bp):
DNA Shearing - M.magister gDNA Shearing All Samples and Bioanalyzer
I previously ran some shearing tests on 20201022 to determine how many cycles to run on the sonicator (Bioruptor 300; Diagenode) to achieve an average fragment length of ~350 - 500bp in preparation for MBD-BSseq. The determination was 70 cycles (30s ON, 30s OFF; low intensity), sonicating for 35 cycles, followed by successive rounds of 5 cycles each.
DNA Shearing - M.magister CH05-21 gDNA Full Shearing Test and Bioanalyzer
Yesterday, I did some shearing of Metacarcinus magister gill gDNA on a test sample (CH05-21) to determine how many cycles to run on the sonicator (Bioruptor 300; Diagenode) to achieve an average fragment length of ~350 - 500bp in preparation for MBD-BSseq. The determination from yesterday was 70 cycles (30s ON, 30s OFF; low intensity). That determination was made by first sonicating for 35 cycles, followed by successive rounds of 5 cycles each. I decided to repeat this, except by doing it in a single round of sonication.
DNA Shearing - M.magister gDNA Shear Testing and Bioanalyzer
Steven assigned me to do some MBD-BSseq library prep (GitHub Issue) for some Dungeness crab (Metacarcinus magister) DNA samples provided by Mackenzie Gavery. The DNA was isolated from juvenile (J6/J7 developmental stages) gill tissue. One of the first steps in MBD-BSseq is to fragment DNA to a desired size (~350 - 500bp in our case). However, we haven’t worked with Metacarcinus magister DNA previously, so I need to empirically determine sonicator (Bioruptor 300; Diagenode) settings for these samples.
Read Mapping - C.bairdi 201002558-2729-Q7 and 6129-403-26-Q7 Taxa-Specific NanoPore Reads to cbai_genome_v1.01.fasta Using Minimap2 on Mox
After extracting FastQ reads using seqtk on 20201013 from the various taxa I had been interested in, the next thing needed doing was mapping reads to the cbai_genome_v1.01 “genome” assembly from 20200917. I found that Minimap2 will map long reads (e.g. NanoPore), in addition to short reads, so I decided to give that a rip.
Data Wrangling - C.bairdi NanoPore Reads Extractions With Seqtk on Mephisto
In my pursuit to identify which contigs/scaffolds of our “C.bairdi” genome assembly from 20200917 correspond to interesting taxa, based on taxonomic assignments produced by MEGAN6 on 20200928, I used MEGAN6 to extract taxa-specific reads from cbai_genome_v1.01 on 20201007 - the output is only available in FastA format. Since I want the original reads in FastQ format, I will use the FastA sequence IDs (from the FastA index file) and provide that to seqtk to extract the FastQ reads for each sample and corresponding taxa.
NanoPore Reads Extractions - C.bairdi Taxonomic Reads Extractions with MEGAN6 on 201002558-2729-Q7 and 6129-403-26-Q7
After completing the taxonomic comparisons of 201002558-2729-Q7 and 6129-403-26-Q7 on 20201002, I decided to extract reads assigned to the following taxa for further exploration (primarily to identify contigs/scaffolds in our cbai_genome_v1.0.fasta (19MB).
Comparison - C.bairdi 20102558-2729 vs. 6129-403-26 NanoPore Taxonomic Assignments Using MEGAN6
After noticing that the initial MEGAN6 taxonomic assignments for our combined C.bairdi NanoPore data from 20200917 revealed a high number of bases assigned to E.canceri and Aquifex sp., I decided to explore the taxonomic breakdown of just the individual samples to see which of the samples was contributing to these taxonomic assignments most.
Taxonomic Assignments - C.bairdi 6129-403-26-Q7 NanoPore Reads Using DIAMOND BLASTx on Mox and MEGAN6 daa2rma on emu
After noticing that the initial MEGAN6 taxonomic assignments for our combined C.bairdi NanoPore data from 20200917 revealed a high number of bases assigned to E.canceri and Aquifex sp., I decided to explore the taxonomic breakdown of just the individual samples to see which of the samples was contributing to these taxonomic assignments most.
Taxonomic Assignments - C.bairdi 20102558-2729-Q7 NanoPore Reads Using DIAMOND BLASTx on Mox and MEGAN6 daa2rma on emu
After noticing that the initial MEGAN6 taxonomic assignments for our combined C.bairdi NanoPore data from 20200917 revealed a high number of bases assigned to E.canceri and Aquifex sp., I decided to explore the taxonomic breakdown of just the individual samples to see which of the samples was contributing to these taxonomic assignments most.
Data Wrangling - C.bairdi NanoPore 6129-403-26 Quality Filtering Using NanoFilt on Mox
Last week, I ran all of our Q7-filtered C.baird NanoPore reads through MEGAN6 to evaluate the taxonomic breakdown (on 20200917) and noticed that there were a large quantity of bases assigned to E.canceri (a known microsporidian agent of infection in crabs) and Aquifex sp. (a genus of thermophylic bacteria), in addition to the expected Arthropoda assignments. Notably, Alveolata assignments were remarkably low.
Data Wrangling - C.bairdi NanoPore 20102558-2729 Quality Filtering Using NanoFilt on Mox
Last week, I ran all of our Q7-filtered C.baird NanoPore reads through MEGAN6 to evaluate the taxonomic breakdown (on 20200917) and noticed that there were a large quantity of bases assigned to E.canceri (a known microsporidian agent of infection in crabs) and Aquifex sp. (a genus of thermophylic bacteria), in addition to the expected Arthropoda assignments. Notably, Alveolata assignments were remarkably low.
Assembly Assessment - BUSCO C.bairdi Genome v1.01 on Mox
After creating a subset of the cbai_genome_v1.0 of contigs >100bp yesterday (subset named cbai_genome_v1.01), I wanted to generate BUSCO scores for cbai_genome_v1.01. This is primarily just to keep info consistent on our Genomic Resources wiki, as I don’t expect these scores to differ at all from the cbai_genome_v1.0 BUSCO scores.
Data Wrangling - Subsetting cbai_genome_v1.0 Assembly with faidx
Previously assembled cbai_genome_v1.0.fasta with our NanoPore Q7 reads on 20200917 and noticed that there were numerous sequences that were well shorter than the expected 500bp threshold that the assembler (Flye) was supposed to spit out. I created an Issue on the Flye GitHub page to find out why. The developer responded and determined it was an issue with the assembly polisher and that sequences <500bp could be safely ignored.
SRA Submissions - NanoPore C.bairdi 20102558-2729 and 6129_403_26
Submitted our C.bairdi NanoPore sequencing data from 20200109 (Sample 20102558-2729 - uninfected EtOH-preserved muscle) and from 20200311 (Sample 6129403_26 - RNAlater-preserved _Hematodinium-infected hemolymph) to the NCBI Sequencing Read Archive(SRA).
Assembly Assessment - BUSCO C.bairdi Genome v1.0 on Mox
After using Flye to perform a de novo assembly of our Q7 filtered NanoPore sequencing data on 20200917, I decided to check the “completeness” of the assembly using BUSCO on Mox.
Data Wrangling - C.bairdi NanoPore Quality Filtering Using NanoFilt on Mox
I previously converting our C.bairdi NanoPre sequencing data from the raw Fast5 format to FastQ format for our three sets of data:
Genome Assembly - C.bairdi - cbai_v1.0 - Using All NanoPore Data With Flye on Mox
After quality filtering the C.bairdi NanoPore data earlier today, I performed a de novo assembly using Flye on Mox.
Taxonomic Assignments - C.bairdi NanoPore Reads Using DIAMOND BLASTx on Mox and MEGAN6 daa2rma on swoose
Earlier today I quality filtered (>=Q7) our C.baird NanoPore reads. One of the things I’d like to do now is to attempt to filter reads taxonomically, since the NanoPore data came from both an uninfected crab and Hematodinium-infected crab.
qPCR - Geoduck Normalizing Gene Primers 28s-v4 and EF1a-v1 Tests
On Monday (20200914), I checked a set of 28s and EF1a primer sets and determined that 28s-v4 and EF1a-v1 were probably the best of the bunch, although they all looked great. So, I needed to test these out on some individual cDNA samples to see if they might be useful as normalizing genes - should have consistent Cq values across all samples/treatments.
qPCR - Geoduck Normalizing Gene Primer Checks
Shelly ordered some new primers (designed by Sam Gurr) (GitHub Issue) to potentially use as normalizing genes for her geoduck reproduction gene expression project and asked that I test them out.
Data Wrangling - Visualization of C.bairdi NanoPore Sequencing Using NanoPlot on Mox
I previously converting our C.bairdi NanoPre sequencing data from the raw Fast5 format to FastQ format for our three sets of data:
Data Wrangling - NanoPore Fast5 Conversion to FastQ of C.bairdi 6129_403_26 on Mox with GPU Node
Time to start working with the NanoPore data that I generated back in March (???!!!). In order to proceed, I first need to convert the raw Fast5 files to FastQ. To do so, I’ll use the NanoPore program guppy.
Data Wrangling - NanoPore Fast5 Conversion to FastQ of C.bairdi 20102558-2729 Run-02 on Mox with GPU Node
Continuing to work with the NanoPore data that I generated back in January(???!!!). In order to proceed, I first need to convert the raw Fast5 files to FastQ. To do so, I’ll use the NanoPore program guppy. I converted the first run from this flowcell earlier today.
Data Wrangling - NanoPore Fast5 Conversion to FastQ of C.bairdi 20102558-2729 Run-01 on Mox with GPU Node
Time to start working with the NanoPore data that I generated back in January(???!!!). In order to proceed, I first need to convert the raw Fast5 files to FastQ. To do so, I’ll use the NanoPore program guppy.
Samples Submission - Supplemental Ronits C.gigas Diploid-Triploid Ctendidia gDNA for WGBS by ZymoResearch
I re-quantified samples for Zymo whole genome bisulfite sequencing (WGBS) project 3534 earlier today due to ZymoResearch indicating there was insufficient DNA for most of the samples and submitted an additional 1ug of each sample. See table below for volume of DNA sent based on today’s (20200901) quants:
DNA Quantification - Re-quant Ronits C.gigas Diploid-Triploid Ctenidia gDNA Submitted to ZymoResearch
I received notice from ZymoResearch yesterday afternoon that the DNA we sent on 20200820 for this project (Quote 3534) had insufficient DNA for sequencing for most of the samples. This was, honestly, shocking. I had even submitted well over the minimum amount of DNA required (submitted 1.75ug - only needed 1ug). So, I’m not entirely sure what happened here.
Transcriptome Annotation - Trinotate C.bairdi v2.1 on Mox
To continue annotation of our C.bairdi v2.1 transcriptome assembly], I wanted to run Trinotate.
Transcriptome Annotation - Trinotate C.bairdi v3.1 on Mox
To continue annotation of our C.bairdi v3.1 transcriptome assembly], I wanted to run Trinotate.
Transcriptome Annotation - Trinotate Hematodinium v3.1 on Mox
Transcriptome Annotation - Trinotate Hematodinium v2.1 on Mox
TransDecoder - C.bairdi Transcriptomes v2.1 and v3.1 on Mox
To continue annotation of our C.bairdi v2.1 & v3.1 transcriptome assemblies, I needed to run TransDecoder before performing the more thorough annotation with Trinotate.
qPCR - P.generosa RPL5 and TIF3s6b v2 and v3 Normalizing Gene Assessment
After testing out the RPL5 and TIF3s6b v2 and v3 primers yesterday on pooled cDNA, we determined the primers looked good, so will go forward testing them on a set of P.generosa hemolymph cDNA made by Kaitlyn on 20200212. This will evaluate whether or not these can be utilized as normalizing genes for subsequent gene expression analyses.
Sample Submitted - C.gigas Diploid-Triploid pH Treatments Ctenidia to ZymoResearch for WGBS
Submitted 1.5ug of the 24 C.gigas ctenidia ctenidia gDNA isolated last week (20200821) to ZymoResearch for whole genome bisulfite sequencing (WGBS) to compare differences in diploid/triploids and responses to elevated pH:
qPCR - P.generosa RPL5-v2-v3 and TIF3s6b-v2-v3 Primer Tests
Shelly ordered some new primers as potential normalizing genes and asked me to test them out (GitHub Issue).
DNA Isolation and Quantification - C.gigas High-Low pH Triploid and Diploid Ctenidia
Isolated DNA from 24 of the Crassostrea gigas high/low pH triploid/diploid ctenidia samples that we received yesterday from the Haws Lab. Samples selected by Steven.
Samples Received - C.gigas High-Low pH Triploid Diploid from Maria Haws Lab
Received Crassostrea gigas tissue samples from Maria Haws’ Lab at the University of Hawaii Hilo. Tissue samples are a set of triploid and diploid subjected to different pHs. All sample info is in the sample sheet provided below.
Samples Submitted - Ronits C.gigas Diploid and Triploid Ctenidia to ZymoResearch for WGBS
Submitted 1.75ug of gDNA from 10 Crassostrea gigas ctenidia samples from Ronit’s dessication/temp/ploidy experiment to ZymoResearch for whole genome bisulfite sequencing (BSseq). They will sequence to ~30x coverage, using 150bp PE reads.
Assembly Stats - C.bairdi Transcriptomes v2.1 and v3.1 Trinity Stats on Mox
Realized that transcriptomes v2.1 and v3.1 (extracted from BLASTx-annotated FastAs from 20200605) didn’t have any associated stats.
Trimming-FastQC-MultiQC - Robertos C.gigas WGBS FastQ Data with fastp FastQC and MultiQC on Mox
Steven asked me to trim Roberto’s C.gigas whole genome bisulfite sequencing (WGBS) reads (GitHub Issue) “following his methods”. The only thing specified is trimming Illumina adaptors and then trimming 10bp from the 5’ end of reads. No mention of which software was used.
TransDecoder - Hematodinium Transcriptomes v1.6, v1.7, v2.1 and v3.1 on Mox
To continue annotation of our Hematodinium v1.6, v1.7, v2.1 & v3.1 transcriptome assemblies, I needed to run TransDecoder before performing the more thorough annotation with Trinotate.
Assembly Stats - cbaiodinium Transcriptomes v2.1 and v3.1 Trinity Stats on Mox
Working on dealing with our various cbaiodinium sp. transcriptomes and realized that transcriptomes v2.1 and v3.1 (extracted from BLASTx-annotated FastAs from 20200605) didn’t have any associated stats.
Transcriptome Assessment - BUSCO Metazoa on Hematodinium v1.6 v1.7 v2.1 and v3.1 on Mox
Transcriptome Annotation - Hematodinium Transcriptomes v1.6 v1.7 v2.1 v3.1 with DIAMOND BLASTx on Mox
Needed to annotate the Hematodinium sp. transcriptomes that I’ve assembled using DIAMOND BLASTx. This will also be used for additional downstream annotation (TransDecoder, Trinotate):
qPCR - P.generosa APLP and TIF3s8-1 with cDNA
Shelly asked me to run some qPCRs (GitHub Issue), after some of the qPCR results I got from primer tests with normalzing genes and potential gene targets.
FastQ Read Alignment and Quantification - P.generosa Water Metagenomic Libraries to MetaGeneMark Assembly with Hisat2 on Mox
Continuing working on the manuscript for this data, Emma wanted the number of reads aligned to each gene. I previously created and assembly with genes/proteins using MetaGeneMark on 20190103, but the assemby process didn’t output any sort of stastics on read counts.
Primer Design and In-Silico Testing - Geoduck Reproduction Primers
Shelly asked that I re-run the primer design pipeline that Kaitlyn had previously run to design a set of reproduction-related qPCR primers. Unfortunately, Kaitlyn’s Jupyter Notebook wasn’t backed up and she accidentally deleted it, I believe, so there’s no real record of how she designed the primers. However, I do know that she was unable to run the EMBOSS primersearch tool, which will check your primers against a set of sequences for any other matches. This is useful for confirming specificity.
qPCR - Testing P.generosa Reproduction-related Primers
Ran some qPCRs on some other primers on 20200723 and then Shelly has asked me to test some additional qPCR primers that might have acceptable melt curves and be usable as normalizing genes.
SRA Submission - P.generosa Metagenomics Data
Added our P.generosa metagenomics sequencing data to NCBI sequencing read archive (SRA).
qPCR - Testing P.generosa Reproduction-related Primers
Shelly has asked me to test some qPCR primers related to geoduck reproduction.
DNA Isolation and Quantification - C.gigas Diploid (Ronit) and Triploid (Nisbet)
Isolated some gDNA from the triploid Nisbet oysters we received on 20200218 and one of Ronit’s diploid ctenidia samples (Google Sheet) using the E.Z.N.A. Mollusc DNA Kit (Omega). See the “Results” section for sample info.
SRA Submission - Geoduck Epigenetic Ocean Acidification RNAseq
Can’t remember where it was discussed (probably lab meeting), but I created a GitHub Issue to add all of geoduck RNAseq data to NCBI Short Read Archive (SRA). Anyway, got all the remaining RNAseq data uploaded to the NCBI SRA and organized into the correct BioSamples and BioProjects.
ENA Submission - Ostrea lurida draft genome Olurida_v081.fa
Submitted our Ostrea lurida v081 genome assembly FastA to the European Nucloetide Archive.
Metagenomics - Data Extractions Using MEGAN6
Decided to finally take the time to methodically extract data from our metagenomics project so that I have the tables handy when I need them and I can easily share them with other people. Previously, I hadn’t done this due to limitations on looking at the data remotely. I finally downloaded all of the RMA6 files from 20191014 after being fed up with the remote desktop connection and upgrading the size of my hard drive (5 of the six RMA6 files are >40GB in size).
Transcriptome Annotation - C.bairdi Transcriptomes v2.1 and v3.1 Using DIAMOND BLASTx on Mox
Decided to annotate the two C.bairdi transcriptomes , cbai_transcriptome_v2.1 and cbai_transcriptome_v3.1, generated on 20200605 using DIAMOND BLASTx on Mox.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi Transcriptome v3.1
Continuing to try to identify the best C.bairdi transcriptome, we decided to extract all non-dinoflagellate sequences from cbai_transcriptome_v2.0 (RNAseq shorthand: 2018, 2019, 2020-GW, 2020-UW) and cbai_transcriptome_v3.0 (RNAseq shorthand: 2018, 2019, 2020-UW).
Transcriptome Assessment - BUSCO Metazoa on C.bairdi Transcriptome v2.1
Continuing to try to identify the best C.bairdi transcriptome, we decided to extract all non-dinoflagellate sequences from cbai_transcriptome_v2.0 (RNAseq shorthand: 2018, 2019, 2020-GW, 2020-UW) and cbai_transcriptome_v3.0 (RNAseq shorthand: 2018, 2019, 2020-UW).
Sequence Extractions - C.bairdi Transcriptomes v2.0 and v3.0 Excluding Alveolata with MEGAN6 on Swoose
Continuing to try to identify the best C.bairdi transcriptome, we decided to extract all non-dinoflagellate sequences from cbai_transcriptome_v2.0 (RNAseq shorthand: 2018, 2019, 2020-GW, 2020-UW) and cbai_transcriptome_v3.0 (RNAseq shorthand: 2018, 2019, 2020-UW). Both of these transcriptomes were assembled without any taxonomic filter applied. DIAMOND BLASTx and conversion to MEGAN6 RMA6 files was performed yesterday (20200604).
Transcriptome Annotation - C.bairdi Transcriptomes v2.0 and v3.0 with DIAMOND BLASTx on Mox
Continuing to try to identify the best C.bairdi transcriptome, we decided to extract all non-dinoflagellate sequences from cbai_transcriptome_v2.0 (RNAseq shorthand: 2018, 2019, 2020-GW, 2020-UW) and cbai_transcriptome_v3.0 (RNAseq shorthand: 2018, 2019, 2020-UW). Both of these transcriptomes were assembled without any taxonomic filter applied.
Transcriptome Comparison - C.bairdi Transcriptomes Evaluations with DETONATE on Mox
Transcriptome Comparison - C.bairdi Transcriptomes Compared with DETONATE on Mox
We’ve produced a number of C.bairdi transcriptomes and we’re interested in doing some comparisons to try to determine which one might be “best”. I previously compared the BUSCO scores of each of these transcriptomes and now will be using the DETONATE software package to perform two different types of comparisons: compared to a reference (REF-EVAL) and determine an overall quality “score” (RSEM-EVAL). I’ll be running REF-EVAL in this notebook.
Transcriptome Annotation - Trinotate C.bairdi Transcriptome-v1.7 on Mox
After creating a de novo assembly of C.bairdi transcriptome v1.7 on 20200527, performing BLASTx annotation on 202000527, and TransDecoder for ORF identification on 20200527, I continued the annotation process by running Trinotate.
Transcriptome Comparisons - C.bairdi BUSCO Scores
Since we’ve generated a number of versions of the C.bairdi transcriptome, we’ve decided to compare them using various metrics. Here, I’ve compared the BUSCO scores generated for each transcriptome using BUSCO’s built-in plotting script. The script generates a stacked bar plot of all BUSCO short summary files that it is provided with, as well as the R code used to generate the plot.
TransDecoder - C.bairdi Transcriptome v1.7 on Mox
Need to run TransDecoder on Mox on the C.bairdi transcriptome v1.7 from 20200527.
Transcriptome Annotation - C.bairdi Transcriptome v1.7 Using DIAMOND BLASTx on Mox
As part of annotating cbai_transcriptome_v1.7.fasta from 20200527, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi Transcriptome v1.7
I previously created a C.bairdi de novo transcriptome assembly v1.7 with Trinity from all our C.bairdi taxonomically filtered pooled RNAseq samples on 20200527 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Assembly - C.bairdi All Pooled Arthropoda-only RNAseq Data with Trinity on Mox
For completeness sake, I wanted to create an additional C.bairdi transcriptome assembly that consisted of Arthropoda only sequences from just pooled RNAseq data (since I recently generated a similar assembly without taxonomically filtered reads on 20200518). This constitutes samples we have designated: 2018, 2019, 2020-UW. A de novo assembly was run using Trinity on Mox. Since all pooled RNAseq libraries were stranded, I added this option to Trinity command.
Transcriptome Annotation - Trinotate C.bairdi Transcriptome-v3.0 on Mox
After performing de novo assembly on all of our Tanner crab RNAseq data (no taxonomic filter applied, either) on 20200518, I continued the annotation process by running Trinotate.
Transcriptome Assembly - P.trituberculatus (Japanese blue crab) NCBI SRA BioProject PRJNA597187 Data with Trinity on Mox
After generating a number of C.bairdi (Tanner crab) transcriptomes, we decided we should compare them to evaluate which to help decide which one should become our “canonical” version. As part of that, the Trinity wiki offers a list of tools that one can use to check the quality of transcriptome assemblies. Some of those require a transcriptome of a related species.
SRA Library Assessment - Determine RNAseq Library Strandedness from P.trituberculatus SRA BioProject PRJNA597187
We’ve produced a number of C.bairid transcriptomes utilizing different assembly approaches (e.g. Arthropoda reads only, stranded libraries only, mixed strandedness libraries, etc) and we want to determine which of them is “best”. Trinity has a nice list of tools to assess the quality of transcriptome assemblies, but most of the tools rely on comparison to a transcriptome of a related species.
Tutorial - SRA Toolkit for Data Retrieval and Conversion to FastQ
I was looking for some crab transcriptomic data today and, unable to find any previously assembled transcriptomes, turned to the good ol’ NCBI SRA. In order to simplify retrieval and conversion of SRA data, need to use the SRA Toolkit software suite. Since I haven’t used this in many years, I figured I might as well put together a brief guide/tutorial so I can refer back to it in the future.
Transcriptome Annotation - Trinotate C.bairdi Transcriptome-v1.6 on Mox
After creating a de novo assembly of C.bairdi transcriptome v1.6 on 20200518, performing BLASTx annotation on 202000519, and TransDecoder for ORF identification on 20200519, I continued the annotation process by running Trinotate.
TransDecoder - C.bairdi Transcriptome v1.6 on Mox
Need to run TransDecoder on Mox on the C.bairdi transcriptome v1.6 from 20200518.
TransDecoder - C.bairdi Transcriptome v3.0 from 20200518 on Mox
Need to run TransDecoder on Mox on the C.bairdi transcriptome v3.0 from 20200518.
Transcriptome Annotation - C.bairdi Transcriptome v1.6 Using DIAMOND BLASTx on Mox
As part of annotating cbai_transcriptome_v1.6.fasta from 20200518, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi Transcriptome v1.6
I previously created a C.bairdi de novo transcriptome assembly v1.6 with Trinity from all our C.bairdi taxonomically filtered RNAseq on 20200518 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Annotation - C.bairdi Transcriptome v3.0 Using DIAMOND BLASTx on Mox
As part of annotating cbai_transcriptome_v3.0.fasta from 20200518, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi Transcriptome v3.0
I previously created a C.bairdi de novo transcriptome assembly with Trinity from all our C.bairdi pooled RNAseq (not taxonomically filtered) on 20200518 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Assembly - C.bairdi All Arthropoda-specific RNAseq Data with Trinity on Mox
I realized I hadn’t performed taxonomic read separation from one set of RNAseq data we had. And, since I was on a transcriptome assembly kick, I figured I’d generate another C.bairdi transcriptome that included only Arthropoda-specific sequence data from all of our RNAseq.
Data Wrangling - Arthropoda and Alveolata D26 Pool RNAseq FastQ Extractions
After using MEGAN6 to extract Arthropoda and Alveolata reads from our RNAseq data on 20200114, I had then extracted taxonomic-specific reads and aggregated each into basic Read 1 and Read 2 FastQs to simplify transcriptome assembly for C.bairdi and for Hematodinium. That was fine and all, but wasn’t fully thought through.
Transcriptome Assembly - C.bairdi All Pooled RNAseq Data Without Taxonomic Filters with Trinity on Mox
Steven asked that I assemble a transcriptome with just our pooled C.bairdi RNAseq data (not taxonomically filtered; see the FastQ list file linked in the Results section below). This constitutes samples we have designated: 2018, 2019, 2020-UW. A de novo assembly was run using Trinity on Mox. Since all pooled RNAseq libraries were stranded, I added this option to Trinity command.
Transcriptome Annotation - Trinotate C.bairdi Transcriptome v2.0 from 20200502 on Mox
After performing de novo assembly on all of our Tanner crab RNAseq data (no taxonomic filter applied, either) on 20200502 and performing BLASTx annotation on 20200508, I continued the annotation process by running Trinotate.
TransDecoder - C.bairdi Transcriptome v2.0 from 20200502 on Mox
Need to run TransDecoder on Mox on the C.bairdi transcriptome v2.0 from 20200502.
Transcriptome Annotation - C.bairdi Transcriptome v2.0 Using DIAMOND BLASTx on Mox
As part of annotating the C.bairdi v2.0 transcriptome assembly from 20200502, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi v2.0 Transcriptome
I previously created a C.bairdi de novo transcriptome assembly with Trinity using all existing, unfiltered (i.e. no taxonomic selection) RNAseq data on 20200502 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Assembly - C.bairdi All RNAseq Data Without Taxonomic Filters with Trinity on Mox
Steven asked that I assemble an unfiltered (i.e. no taxonomic selection) transcriptome with all of our C.bairdi RNAseq data (see the FastQ list file linked in the Results section below). A de novo assembly was run using Trinity on Mox. It should be noted that this assembly is a mixture of stranded/non-stranded library preps.
Transcript Abundance - C.bairdi Alignment-free with Salmon Using 2020-GW Data on Mox
Clarified with Steven an approach for tackling multi-condition comparisons (see this GitHub Issue). As such, I need to have individual transcript abundances for each sample from the 2020 Genewiz RNAseq data before I can proceed. So, I ran salmon (v1.2.1) to perform an alignment-free set of transcript abundances. It’s ridiculously fast, btw…
GO to GOslim - C.bairdi Enriched GO Terms from 20200422 DEGs
After running pairwise comparisons and identify differentially expressed genes (DEGs) on 20200422 and finding enriched gene ontology terms, I decided to map the GO terms to Biological Process GOslims. Additionally, I decided to try another level of comparison (I’m not sure how valid it is), whereby I will count the number of GO terms assigned to each GOslim and then calculate the percentage of GOterms that get assigned to each of the GOslim categories. The idea being that it might help identify Biological Processes that are “favored” in a given set of DEGs. I decided to set up “fancy” pyramid plots to view a given set of GO-GOslims for each DEG comparison.
FastQC-MultiQC - Laura Spencer’s QuantSeq Data
Laura Spencer received her O.lurida QuantSeq data, so I put it through FastQC/MultiQC and put the pertinent info in the nightingales Google Sheet. I also moved the data to /owl/nightingales/O_lurida, updated the readme file and checksums file. There were 148 individual samples, so I won’t list them all here.
Gene Expression - C.bairdi Pairwise DEG Comparisons with 2019 RNAseq using Trinity-Salmon-EdgeR on Mox
Per a Slack request, Steven asked me to take the Genewize RNAseq data (received 2020318) through edgeR. Ran the analysis using the Trinity differential expression pipeline:
RNAseq Reads Extractions - C.bairdi Taxonomic Reads Extractions with MEGAN6 on swoose
Transcript Abundance - C.bairdi Alignment-free with Salmon on Mox for Grace
Per this GitHub Issue, Grace and Steven asked if I could help by generating a transcript abundance file for Grace to use with EdgeR. To do so, I used Salmon for alignment-free transcript abundance estimates due to its speed and its incorporation into Trinity with the following files:
SRA Submission - C.bairdi RNAseq Data
Since we received the last of our RNAseq data for this project on 20200413, I submitted all of it to the NCBI Sequencing Read Archive (SRA). Data was released today and all accession numbers can be found in the table below:
TrimmingFastQCMultiQC—C.bairdi-RNAseq-FastQ-with-fastp-on-Mox
After receiving our RNAseq data from Genewiz earlier today, needed to trim and check trimmed reads with FastQC.
Taxonomic Assignments - C.bairdi RNAseq Using DIAMOND BLASTx on Mox and MEGAN6 Meganizer on swoose
After receiving/trimming the latest round of C.bairdi RNAseq data on 20200413, need to get the data ready to perform taxonomic selection of sequencing reads. To do this, I first need to run DIAMOND BLASTx, then “meganize” the output files in preparation for loading into MEGAN6, which will allow for taxonomic-specific read separation.
FastQC-MultiQC - C.bairdi Raw RNAseq from NWGSC
Yesterday, we received the last of the RNAseq data for the C.bairdi crab project from NWGSC. FastQC, followed by MultiQC was run on the raw FastQ reads on my computer (swoose).
Data Wrangling - Arthropoda and Alveolata Day and Treatment Taxonomic RNAseq FastQ Extractions
After using MEGAN6 to extract Arthropoda and Alveolata reads from our RNAseq data on 20200330, I had then extracted taxonomic-specific reads and aggregated each into basic Read 1 and Read 2 FastQs to simplify transcriptome assembly for C.bairdi and for Hematodinium. That was fine and all, but wasn’t fully thought through.
Data Received - C.bairdi RNAseq from NWGSC
Received the C.bairdi RNAseq data from UW NWGSC that Grace submitted on 20200131. Below is a table of NWGSC sample name and corresponding labels that Grace provided. The 0/1 indicates uninfected/infected, respectively.
Transcriptome Annotation - Trinotate C.bairdi MEGAN6 Taxonomic-specific Trinity Assembly on Mox
After performing de novo assembly on our Tanner crab MEGAN6 taxonomic-specific RNAseq data on 20200330 and performing BLASTx annotation on 20200408, I continued the annotation process by running Trinotate.
Transcriptome Annotation - C.bairdi MEGAN Trinity Assembly Using DIAMOND BLASTx on Mox
As part of annotating the most recent transcriptome assembly from the MEGAN6 Arthropoda taxonomic-specific reads, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Annotation - Trinotate Hematodinium MEGAN6 Taxonomic-specific Trinity Assembly on Mox
After performing de novo assembly on our Hematodinium MEGAN6 taxonomic-specific RNAseq data on 20200330 and performing BLASTx annotation on 20200331, I continued the annotation process by running Trinotate.
Transdecoder - Hematodinium MEGAN6 Taxonomic-Specific Reads Assembly from 20200330
Ran Trinity to de novo assembly on the the Alveolata MEGAN6 taxonomic-specific RNAseq data on 20120330 and now will begin annotating the transcriptome using TransDecoder on Mox.
Transdecoder - C.bairdi MEGAN6 Taxonomic-Specific Reads Assembly from 20200330
Ran Trinity to de novo assembly on the the Arthropoda MEGAN6 taxonomic-specific RNAseq data on 20120330 and now will begin annotating the transcriptome using TransDecoder on Mox.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi MEGAN Transcriptome
I previously created a C.bairdi de novo transcriptome assembly with Trinity from the MEGAN6 taxonomic-specific reads for Arthropoda on 20200330 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Annotation - Hematodinium MEGAN Trinity Assembly Using DIAMOND BLASTx on Mox
As part of annotating the most recent transcriptome assembly from the MEGAN6 Hematodinium taxonomic-specific reads, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Assessment - BUSCO Metazoa on Hematodinium MEGAN Transcriptome
I previously created a C.bairdi de novo transcriptome assembly with Trinity from the MEGAN6 taxonomic-specific reads for Alveolata on 20200331 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Assembly - Hematodinium with MEGAN6 Taxonomy-specific Reads with Trinity on Mox
Ran a de novo assembly using the extracted reads classified under Alveolata from:
Transcriptome Assembly - C.bairdi with MEGAN6 Taxonomy-specific Reads with Trinity on Mox
Ran a de novo assembly using the extracted reads classified under Arthropoda from:
RNAseq Reads Extractions - C.bairdi Taxonomic Reads Extractions with MEGAN6 on swoose
Transcriptome Annotation - C.bairdi Using DIAMOND BLASTx on Mox and MEGAN6 Meganizer on swoose
After receiving/trimming the latest round of C.bairdi RNAseq data on 20200318, need to get the data ready to perform taxonomic selection of sequencing reads. To do this, I first need to run DIAMOND BLASTx, then “meganize” the output files in preparation for loading into MEGAN6, which will allow for taxonomic-specific read separation.
Trimming/FastQC/MultiQC - C.bairdi RNAseq FastQ with fastp on Mox
After receiving our RNAseq data from Genewiz earlier today, needed to run FastQC, trim, check trimmed reads with FastQC.
Data Received - C.bairdi RNAseq Data from Genewiz
We received the RNAseq data from the RNA that was sent out by Grace on 20200212.
DNA Isolation and Quantification - C.bairdi Hemocyte Pellets in RNAlater
Isolated DNA from 22 samples (see Qubit spreadsheet in “Results” below for sample IDs) using the Quick DNA/RNA Microprep Kit (ZymoResearch; PDF) according to the manufacturer’s protocol for liquids/cells in RNAlater.
NanoPore Sequencing - C.bairdi gDNA 6129_403_26
After getting high quality gDNA from Hematodinium-infected C.bairdi hemolymph on 2020210 we decided to run some of the sample on the NanoPore MinION, since the flowcells have a very short shelf life. Additionally, the results from this will also help inform us on whether this sample might worth submitting for PacBio sequencing. And, of course, this provides us with additional sequencing data to complement our previous NanoPore runs from 20200109.
qPCR - C.bairdi RNA Check for Residual gDNA
Previuosly checked existing crab RNA for residual gDNA on 20200226 and identified samples with yields that were likely too low, as well as samples with residual gDNA. For those samples, was faster/easier to just isolate more RNA and perform the in-column DNase treatment in the ZymoResearch Quick DNA/RNA Microprep Plus Kit; this keeps samples concentrated. So, I isolated more RNA on 20200306 and now need to check for residual gDNA.
Trimming/MultiQC - Methcompare Bisulfite FastQs with fastp on Mox
Steven asked me to trim a set of FastQ files, provided by Hollie Putnam, in preparation for methylation analysis using Bismark. The analysis is part of a coral project comparing DNA methylation profiles of different species, as well as comparing different sample prep protocols. There’s a dedicated GitHub repo here:
RNA Isolation and Quantification - C.bairdi RNA from Hemolymph Pellets in RNAlater
Based on qPCR results testing for residual gDNA from 20200225, a set of 24 samples were identified that required DNase treatment and/or additional RNA. I opted to just isolate more RNA from all samples, since the kit includes a DNase step and avoids diluting the existing RNA using the Turbo DNA-free Kit that we usully use. Isolated RNA using the Quick DNA/RNA Microprep Kit (ZymoResearch; PDF) according to the manufacturer’s protocol for liquids/cells in RNAlater.
Data Wrangling - Create Canonical Olurida_v081 Genes FastA
I finally had some time to tackle this GitHub Issue and create a canonical genes FastA file using the MAKER IDs, instead of the original contig IDs from our Olympia oyster genome assembly - https://owl.fish.washington.edu/halfshell/genomic-databank/Olurida_v081.fa (FastA; 1.1GB).
qPCR - C.bairdi RNA Check for Residual gDNA
After deciding on a primer set to use for gDNA detection on 20200225, went ahead and ran a qPCR on most of the RNA samples described in Grace’s Google Sheet. Some samples were not run, as they had not yet been located at the time I began the qPCR.
qPCR - C.bairdi Primer Tests on gDNA
We received the primers I ordered on 20200220 and now need to test them to see if they detect gDNA. If yes, then they’re good candidates to assess the presence of residual gDNA in our RNA samples before we proceed with reverse transcription.
Primer Design - C.bairdi Primers for Checking RNA for Residual gDNA
Getting ready to run some qPCRs and first we need to confirm that our RNA is actually DNA-free. Before we can do that, we need some primers to use, so I decided to semi-arbitrarily select three different gene targets from our MEGAN6 taxonomic-specific Trinity assembly from 20200122.
DNA Isolation & Quantification - C.bairdi RNA from Samples 6212_132_9 6212_334_12 6212_485_26
Isolated DNA from three samples (see Qubit spreadsheet in “Results” below for sample IDs) using the Quick DNA/RNA Microprep Kit (ZymoResearch; PDF) according to the manufacturer’s protocol for liquids/cells in RNAlater.
RNA Isolation & Quantification - C.bairdi RNA from Samples 6212_132_9 6212_334_12 6212_485_26
We are supposed to get RNA sent out for sequencing today, but it turns out that a few of the designated samples have insufficient RNA in them. So, I’m going to attempt to isolate enough RNA from the following samples in order to have enough RNA to send to Genewiz today:
RNA Isolation & Quantification - C.bairdi RNA from Sample 6129_403_26
Since I was isolating gDNA from C.bairdi 6129_403_26 hemolymph, I figured I might as well co-isolate RNA since I was using the Quick DNA/RNA Microprep Plus Kit (ZymoResearch).
DNA Isolation & Quantification - Additional C.bairdi gDNA from Sample 6129_403_26
Earlier today I isolated gDNA from C.bairi 6129_403_26 hemolymph pellets and recovered decently intact gDNA that could be used for sequencing. However, I still need more gDNA, so will isolate that (and co-isolate RNA, since I’m going through the procedure anyway) using the rest of the sample using the Quick DNA/RNA Microprep Plus Kit (ZymoResearch).
DNA Isolation, Quantification, and Gel - C.bairdi gDNA Sample 6129_403_26
In order to do some genome sequencing on C.bairid and Hematodinium, we need hihg molecular weight gDNA. I attempted this twice before, using two different methods (Quick DNA/RNA Microprep Kit (ZymoResearch) on 20200122 and the E.Z.N.A Mollusc DNA Kit (Omega) on 20200108) using ~10yr old ethanol-preserved tissue provided by Pam Jensen. Both methods yielded highly degrade gDNA. So, I’m now attempting to get higher quality gDNA from the RNAlater-preserved hemolymph pellets from this experiment.
Transcriptome Assessment - BUSCO Metazoa on Hematodinium MEGAN Transcriptome
I previously created a Hematodinium de novo transcriptome assembly with Trinity from the MEGAN6 taxonomic-specific reads for Alveolata on 20200122 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Transcriptome Assessment - BUSCO Metazoa on C.bairdi MEGAN Transcriptome
I previously created a C.bairdi de novo transcriptome assembly with Trinity from the MEGAN6 taxonomic-specific reads for Arthropoda on 20200122 and decided to assess its “completeness” using BUSCO and the metazoa_odb9 database.
Gene Expression - Hematodinium MEGAN6 with Trinity and EdgeR
After completing annotation of the Hematodinium MEGAN6 taxonomic-specific Trinity assembly using Trinotate on 20200126, I performed differential gene expression analysis and gene ontology (GO) term enrichment analysis using Trinity’s scripts to run EdgeR and GOseq, respectively. The comparison listed below is the only comparison possible, as there were no reads present in the uninfected Hematodinium extractions.
Gene Expression - C.bairdi MEGAN6 with Trinity and EdgeR
After completing annotation of the C.bairdi MEGAN6 taxonomic-specific Trinity assembly using Trinotate on 20200126, I performed differential gene expression analysis and gene ontology (GO) term enrichment analysis using Trinity’s scripts to run EdgeR and GOseq, respectively, across all of the various treatment comparisons. The comparison are listed below and link to each individual SBATCH script (GitHub) used to run these on Mox.
Data Wrangling - Arthropoda and Alveolata Day and Treatment Taxonomic RNAseq FastQ Extractions
After using MEGAN6 to extract Arthropoda and Alveolata reads from our RNAseq data on 20200114, I had then extracted taxonomic-specific reads and aggregated each into basic Read 1 and Read 2 FastQs to simplify transcriptome assembly for C.bairdi and for Hematodinium. That was fine and all, but wasn’t fully thought through.
DNA Isolation and Quantification - C.bairdi Hemocyte Pellets in RNAlater
Isolated DNA from 56 samples (see Qubit spreadsheet in “Results” below for sample IDs) using the Quick DNA/RNA Microprep Kit (ZymoResearch; PDF) according to the manufacturer’s protocol for liquids/cells in RNAlater.
Transcriptome Annotation - Trinotate Hematodinium MEGAN6 Taxonomic-specific Trinity Assembly on Mox
After performing de novo assembly on our Hematodinium MEGAN6 taxonomic-specific RNAseq data on 20200122 and performing BLASTx annotation on 20200123, I continued the annotation process by running Trinotate.
Transcriptome Annotation - Trinotate C.bairdi MEGAN6 Taxonomic-specific Trinity Assembly on Mox
After performing de novo assembly on our Tanner crab MEGAN6 taxonomic-specific RNAseq data on 20200122 and performing BLASTx annotation on 20200123, I continued the annotation process by running Trinotate.
RNA Isolation and Quantification - C.bairdi Hemocyte Pellets in RNAlater
Isolated RNA from the following hemolymph pellet samples:
RNA Isolation and Quantification - C.bairdi Hemocyte Pellets in RNAlater
Isolated RNA from the following hemolymph pellet samples:
Transdecoder - Hematodinium MEGAN6 Taxonomic-Specific Reads Assembly from 20200122
Ran Trinity to de novo assembly on the the C.bairdi MEGAN6 taxonomic-specific RNAseq data on 201200122 and now will begin annotating the transcriptome using TransDecoder on Mox.
Transdecoder - C.bairdi MEGAN6 Taxonomic-Specific Reads Assembly from 20200122
Ran Trinity to de novo assembly on the the Hematodinium MEGAN6 taxonomic-specific RNAseq data on 201200122 and now will begin annotating the transcriptome using TransDecoder on Mox.
Transcriptome Annotation - Hematodinium MEGAN Trinity Assembly Using DIAMOND BLASTx on Mox
As part of annotating the transcriptome assembly from the MEGAN6 Hematodinium taxonomic-specific reads, I need to run DIAMOND BLASTx to use with Trinotate.
Transcriptome Annotation - C.bairdi MEGAN Trinity Assembly Using DIAMOND BLASTx on Mox
As part of annotating the transcriptome assembly from the MEGAN6 C.bairdi taxonomic-specific reads, I need to run DIAMOND BLASTx to use with Trinotate.
RNA Isolation and Quantification - C.bairdi Hemocyte Pellets in RNAlater Troubleshooting
After the failure to obtain RNA from any C.bairdi hemocytes pellets (out of 24 samples processed) on 20200117, I decided to isolate RNA from just a subset of that group to determine if I screwed something up last time or something. Also, I am testing two different preparations of the kit-supplied DNase I: one Kaitlyn prepped and a fresh preparation that I made. Admittedly, I’m not doing the “proper” testing by trying the different DNase preps on the same exact sample, but it’ll do. I just want to see if I get some RNA from these samples this time…
DNA Quality Assessment - Agarose Gel for C.bairdi 20102558-2729 gDNA from 20200122
Earlier today, I isolated gDNA from C.bairdi 20102558-2729 ethanol-preserved muscle tissue using the Quick DNA/RNA MicroPrep Plus Kit (ZymoResearch) and prepared the tissue in three different ways to see how they would compare:
Data Wrangling - Arthropoda and Alveolata Taxonomic RNAseq FastQ Extractions
After using MEGAN6 to extract Arthropoda and Alveolata reads from our RNAseq data on 20200114 (for reference, these include RNAseq data using a newly established “shorthand”: 2018, 2019), I realized that the FastA headers were incomplete and did not distinguish between paired reads. Here’s an example:
DNA Isolation - C.bairdi 20102558-2729 EtOH-preserved Tissue via Three Variations Using Quick DNA-RNA MicroPrep Kit
Previously, I isolated gDNA from a C.bairdi EtOH-preserved muscle sample (20102558-2729) on 20200108 using the E.Z.N.A. Mollusc DNA Kit (Omega). Although the yields were excellent, the DNA looked completely degraded on a gel and running that DNA on a minION flowcell yielded relatively short reads (which wasn’t terribly surprising).
Transcriptome Assembly - Hematodinium with MEGAN6 Taxonomy-specific Reads with Trinity on Mox
Ran a de novo assembly using the extracted reads classified under Alveolata from 20200122 The assembly was performed with Trinity on Mox.
Transcriptome Assembly - C.bairdi with MEGAN6 Taxonomy-specific Reads with Trinity on Mox
Ran a de novo assembly using the extracted reads classified under Arthropoda from 20200122 (for reference, these include RNAseq data using a newly established “shorthand”: 2018, 2019). The assembly was performed with Trinity on Mox.
DNA Isolation and Quantification - C.bairdi Hemolymph Pellets in RNAlater
Isolated DNA from the following 23 samples:
RNA Isolation and Quantification - C.bairdi Hemolymph Pellets in RNAlater
TL;DR - Recovered absolutely no RNA from any sample! However, I did recover DNA from each sample.
RNAseq Reads Extractions - C.bairdi Taxonomic Reads Extractions with MEGAN6 on swoose
I previously ran BLASTx and “meganized” the output DAA files on 20200103 (for reference, these include RNAseq data using a newly established “shorthand”: 2018, 2019) and now need to use MEGAN6 to bin the results into the proper taxonomies. This is accomplished using the MEGAN6 graphical user interface (GUI). This is how the process goes:
Lab Maintenance - Cluster UPS Battery Replacement
Replaced the batteries on one of the APC uninterruptable power supplies (UPS) on our local server cabinet.
NanoPore Sequencing - C.bairdi gDNA Sample 20102558-2729
I performed the initial Lambda sequencing test on 20200107 and everything went smoothly, so I’m ready to give the NanoPore (ONT) MinION a run with an actual sample!
DNA Quality Assessment - Agarose Gel and NanoDrop on C.bairdi gDNA
I isolated C.bairdi gDNA yesterday (20200108) and now want to get an idea if it’s any good (i.e. no contaminants, high molecule weight).
DNA Isolation and Quantification - C.bairdi gDNA from EtOH Preserved Tissue
I isolated gDNA from ethanol-preserved C.bairdi muscle tissue from sample 20102558-2729 (SPNO-ReferenceNO). This sample was chosen as it had 0 in the SMEAR_result and BCS_PCR_results columns, indicating it should be free of Hematodinium. See the sample spreadsheet linked below for more info.
NanoPore Sequencing - Initial NanoPore MinION Lambda Sequencing Test
We recently acquired a NanoPore MinION sequencer, FLO-MIN106 flow cell and the Rapid Sequencing Kit (SQK-RAD004). The NanoPore website provides a pretty thorough an user-friendly walk-through of how to begin using the system for the first time. With that said, I believe the user needs to have a registered account with NanoPore and needs to have purchased some products to have full access to the protocols they provide.
Transcriptome Annotation - C.bairdi Using DIAMOND BLASTx on Mox and MEGAN6 Meganizer
Although I previously annotated our C.bairdi transcriptome from 20191218, I realized that the assembly and annotations were combine infected/uninfected samples, possibly making separating crab/Hematodinium sequences a bit more difficult.
Reagent Prep - RNA Pico Ladder Aliquoting and Testing
Created aliquots of RNA Pico Ladder received on 20191101, per the manufacturer’s instructions. Created single-use aliquots of 1.5uL and stored at -80oC:
2019
Transcriptome Annotation - C.bairdi Trinity Assembly Trinotate on Mox
After performing de novo assembly on our Tanner crab RNAseq data on 20191218 and performing BLASTx annotation on 20191224, I continued the annotation process by running Trinotate.
Transcriptome Annotation - C.bairdi Trinity Assembly BLASTx on Mox
In preparation for complete transcriptome annotation of the C.bairdi de novo assembly fro 20191218, I needed to run BLASTx. The assembly was BLASTed against the SwissProt database that comes with Trinotate. Initial BLAST output format selected was format 11 (i.e. ASN format), as this allows for simple conversion between different formats later on, if desired.
Transdecoder - C.bairdi De Novo Transcriptome from 20191218 on Mox
Ran Trinity to de novo assemble the C.bairdi RNAseq data we had on 20191218 and now will begin annotating the transcriptome using TransDecoder on Mox.
Transcriptome Assembly - C.bairdi Trimmed RNAseq Using Trinity on Mox
Earlier today, I trimmed our existing C.bairdi RNAseq data, as part of producing generating a transcriptome (per this GitHub issue). After trimming, I performed a de novo assembly using Trinity (v2.9.0) with the stranded library option (--SS_lib_type RF) on Mox.
Trimming/FastQC/MultiQC - C.bairdi RNAseq FastQ with fastp on Mox
Grace/Steven asked me to generate a de novo transcriptome assembly of our current C.bairdi RNAseq data in this GitHub issue. As part of that, I needed to quality trim the data first. Although I could automate this as part of the transcriptome assembly (Trinity has Trimmomatic built-in), I would be unable to view the post-trimming results until after the assembly was completed. So, I opted to do the trimming step separately, to evaluate the data prior to assembly.
Data Wrangling - Olurida_v081 UTR GFFs and Intergenic, Intron BED files
After a meeting last week, we realized we needed to update the paper-oly-mbdbs-gen GitHub repo with the most current versions of feature files we had.
Samples Received - Pacific Oyster Tissues from Hawaii (Maria Haws) from High and Low pCO2
We received frozen Crassostrea gigas tissues from Maria Haws in Hawaii, as part of a pCO2 experiment. Tissues include:
Data Wrangling - Renaming, Splitting, and Feature Counts of Updated Pgenerosa_v074 GenSAS Merged GFF
In the final GFF from our GenSAS Pgenerosa_v074.a4 annotation , we noticed that there were no repeat motifs/sequences identified on Scaffold 01. The remaining scaffolds all had repeat motifs present on them, so something seemed amiss (see this GitHub Issue for more info).
PCR - Crassostrea gigas and sikamea Mantle gDNA from Marinelli Shellfish Company
I ran this PCR a couple of times before and, embarrassingly, I had ordered/used the wrong primers.
Samples Received - C.bairdi Hemolymph and Tissue in Ethanol from Pam Jensen
Pam Jensen (NOAA) brought by a variety of hemolymph and tissues stored in ethanol to use for DNA sequencing with the new MinION (Oxford Nanopore) to help aid our transcriptomics work.
qPCR - Geoduck hemolymph and hemocyte cDNA with vitellogenin primers
Previously isolated RNA on 20191125 and made cDNA on 20191126 from some geoduck hemolymph and hemocyte samples that Shelly asked me to run qPCRs on.
Reverse Transcription - P.generosa DNased Hemolypmh and Hemocyte RNA from 20191125
Performed reverse transcription on the DNased hemolymph and hemocyte RNA from yesterday.
RNA Isolation and Quantification - Geoduck hemolymph and hemocyte samples
Shelly asked me to isolate RNA and run some qPCRs on the following samples:
PCR - Crassostrea gigas and sikamea Mantle gDNA from Marinellie Shellfish Company - No Multiplex
UPDATE 20191125
PCR - Crassostrea gigas and sikamea Mantle gDNA from Marinelli Shellfish Company
UPDATE 20191125
Data Wrangling - Rename Pgenerosa_v074 Bismark Coverage Files Scaffold Names
After a meeting today, Steven and Hollie realized that the Bismark coverage files were still using the old scaffold names, stemming from the Pgenerosa_v074 naming. I’d previously updated filenames and scaffold names, so I recycled some of the code used there to rename the coverage files.
PCR - Crassostrea gigas and sikamea Mantle gDNA from Marinelli Shellfish Company
UPDATE 20191125
DNA Isolation and Quantification - Crassostrea gigas and Crassostrea sikamea Mantle Tissue from Marinelli Shellfish Company
Isolated DNA from the C.gigas and C.sikamea samples we received from Marinelli Shellfish Company on 20191030 using DNAzol.
Data Wrangling - Additional Features Stats for Panopea-generosa-v1.0
Although I’d previously generated some feature stats for this genome annotation (see 20191029), we decided we wanted to get some additional info, similar to that of Table 1 in M.Aranda et al 2016. Scientific Reports.
Data Wrangling - Rename Pgenerosa_v074 Files and Scaffolds
Continuing to organizing files for a manuscript dealing with the geoduck genome assembly/annotation we’ve done, we decided to rename the files as well as rename the scaffolds, to make the naming consistent and a bit easier to read (both for humans and computers).
Data Wrangling - Splitting BAM by Size for Upload to OSF
We’re in the process of organizing files for a manuscript dealing with the geoduck genome assembly/annotation we’ve done. As part of that, we need the Stringtie BAM file that was used with GenSAS for Pgenerosa_v074 annotation to upload to the Open Science Foundation repository for this project. Unfortunately, at 73GB, the file far exceeds the individual file size limit for OSF (5GB). So, I split it into 5GB chunks. See the following notebook for deets:
Samples Received - Marinelli Shellfish Company C.gigas and C.sikamea Oysters
Steven was recently contacted by Marinelli Shellfish Company to see if we could help them determine if some oysters they had were Crassostrea gigas (Pacific oyster) or Crassostrea sikamea (Kumamoto). Steven knows of a paper with primer sequences to use with qPCR for this specific determination.
Data Wrangling - Create Panopea-generosa-vv0.74.a4 Intron and Intergenic BED Files
Since generating an updated Pgenerosa_v074 annotation, we also needed updated intergenic and intron bed files to put in the OSF repository for this project.
Genome Feature Counts - Panopea-generosa-vv0.74.a4
UPDATE 20191203
FastQC-MultiQC - C.bairdi RNAseq Day 12 26 Infected Uninfected
After receiving the rest of the crab data and concatenating it all together, I ran FastQC and MultiQC on the FastQ files.
Data Received - C.bairdi RNAseq Day9-12-26 Infected-Uninfected
Previously, we “received” this data, but it turns out it was incomplete (see 20191003).
Lab Maintenance - Cluster UPS Battery Replacement
Replaced the batteries on one of the APC uninterruptable power supplies (UPS) on our local server cabinet.
Metagenomics Annotation - P.generosa Water Samples with MEGAN6
After running DIAMOND BLASTx and MEGANIZER on these samples on 20190925 to assess taxonomy info, I began the analyses/visualization of this data with MEGAN6.
Data Received - C.bairdi RNAseq Day9-12-26 Infected-Uninfected
UPDATE (20191024): This post details receipt of incomplete data. Additional sequencing was performed and that additional data was received 20191024. This notebook entry on 20191024 contains details on FastQ concatenation of the data below and the data received on 20191024.
Genome Annotation - Pgenerosa_v074 a4 Using GenSAS
I’d previously annotated our Pgenerosa_v074 with GenSAS, but did so using limited options as we were (and still are) in need of an annotated genome to use for methylation data analysis. As such, I opted for speed, but a potentially a less accurate annotation.
Metagenomics Annotation - P.generosa Water Samples Using DIAMOND BLASTx on Mox
Trimming/FastQC/MultiQC - P.generosa EPI FastQs with FASTP on Mox
Steven noticed that the M-Bias plots generated by Bismark from these files was a little wonky and asked that I try trimming them a bit more. The files were originally quality/adaptor trimmed with TrimGalore! on 20180516.
SRA Submission - Hollie’s Juvenile OA BS-seq Data
Submitted sequencing data (52 samples; see table below) from Hollie’s BS-seq project looking at differences in DNA methylation between juvenile geoduck exposed to different pHs over time.
Data Wrangling - Pgenerosa_v074.a3 Annotation Genome Repeats Compostion
Needed to pull some numbers on repeats in our Pgenerosa_v074.a3 GenSAS annotation (from 20190710) for the manuscript we’re putting together.
Data Wrangling - Pgenerosa_v074.a3 Annotation Genome Feature Sequence Lengths
The GenSAS Pgenerosa_v074 annotation from 20190710 (referred to as: Panopea-generosa-vv0.74.a3) recently completed (after nearly a month of running).
Data Wrangling - Panopea generosa Genome Feature Sequence Lengths
In preparation for a paper we’re writing, we needed some summary stats on the various genome assembly feature sequences. I determined the max/min, mean, and median sequence lengths for all the GFF feature files we currently have for Pgenerosa_v070, Pgenerosa_v070_top18_scaffolds, and Pgenerosa_v074. This info will be compiled in to a table for the manuscript. See our Genomic Resources wiki for more info on GFFs:
Data Wrangling - Create a CpG GFF from Pgenerosa_v074 using EMBOSS fuzznuc on Swoose
Steven wanted me to create a CpG GFF for use in IGV visulations for continued Pgenerosa_v074 analysis. I did that by running the EMBOSS tool fuzznuc:
Genome Annotation - Pgenerosa_v074 MAKER on Mox with Stringtie Transcripts GFF
For reference, this annotation will be referred to as Pgenerosa_v074.1.
Genome Comparison - Pgenerosa_v074 vs Pgenerosa_v070 with MUMmer Promer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer 3.23 (specifically, promer for protein level comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs S.glomerata NCBI with MUMmer Promer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer 3.23 (specifically, promer for protein level comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs M.yessoensis NCBI with MUMmer Promer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer 3.23 (specifically, promer for protein level comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs H.sapiens NCBI with MUMmer Promer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer 3.23 (specifically, promer for protein level comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs C.virginica NCBI with MUMmer Promer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer 3.23 (specifically, promer for protein level comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs C.gigas NCBI with MUMmer Promer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer 3.23 (specifically, promer for protein level comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs Pgenerosa_v070 with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs Pgenerosa_v074 with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs S.glomerata NCBI with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs M.yessoensis NCBI with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs H.sapiens NCBI with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs C.gigas NCBI with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
Genome Comparison - Pgenerosa_v074 vs C.virginica NCBI with MUMmer on Mox
In continuing to further improve our geoduck genome annotation, I’m attempting to figure out why Scaffold 1 of our assembly doesn’t have any annotations. As part of that I’ve decided to perform a series of genome comparisons and see how they match up, with an emphasis on Scaffold 1, using MUMmer (v4) (specifically, nucmer for nucleotide comparisons). This software is specifically designed to do this type of comparison.
RepeatMasker - Pgenerosa_v070 for Transposable Element ID on Roadrunner
Continuing our various attempts at annotating our geoduck genome assemblies, I will be re-annotating our Pgenerosa_v070 (see Genome Resources GitHub wiki for deets) and realized I hadn’t run RepeatMasker on this assembly previously. Running RepeatMasker will generate a GFF that I can supply to MAKER to aid in repeats identification.
Data Wrangling - FastA Splitting With faSplit
Steven posted an issue on GitHub regarding splitting a FastA file into multiple sequences. Specifically, he wanted a single, large FastA sequence (~89Mbp) split into smaller FastAs for BLASTing.
Data Summary - P.generosa Transcriptome Assemblies Stats
In our continuing quest to wrangle the geoduck transcriptome assemblies we have, I was tasked with compiling assembly stats for our various assemblies. The table below provides an overview of some stats for each of our assemblies. Links within the table go to the the notebook entries for the various methods from which the data was gathered. In general:
Transcriptome Compression - P.generosa Transcriptome Assemblies Using CD-Hit-est on Mox
In continued attempts to get a grasp on the geoduck transcriptome size, I decided to “compress” our various assemblies by clustering similar transcripts in each assembly in to a single “representative” transcript, using CD-Hit-est. Settings use to run it were taken from the Trinity FAQ regarding “too many transcripts”.
Genome Annotation - Pgenerosa_v074 Transcript Isoform ID with Stringtie on Mox
After annotating Pgenerosa_v070 and comparing feature counts, there was a drastic difference between the two genome versions. Additionally, both of those genomes ended up with no CDS/exon/gene/mRNA features identified in the largest scaffold. So, to explore this further by seeing where (if??) sequencing reads map to the scaffold, and to obtain transcript isoforms for the genome, I ran Stringtie. A Hisat2 index was prepared earlier.
Genome Annotation - Pgenerosa_v070 Transcript Isoform ID with Stringtie on Mox
After annotating Pgenerosa_v074 and comparing feature counts, there was a drastic difference between the two genome versions. Additionally, both of those genomes ended up with no CDS/exon/gene/mRNA features identified in the largest scaffold. So, to explore this further by seeing where (if??) sequencing reads map to the scaffold, and to obtain transcript isoforms for the genome, I ran Stringtie. A Hisat2 index was prepared earlier.
Genome Annotation - Pgenerosa_v074 Hisat2 Transcript Isoform Index
Genome Annotation - Pgenerosa_v070 Hisat2 Transcript Isoform Index
This is the first step in getting transcript isoform annotations. The annotations and alignments that will be generated with Stringtie will be used to help us get a better grasp of what’s going on with our annotations of the different Panopea generosa genome assembly versions.
Genome Annotation - Pgenerosa_v070 and v074 Top 18 Scaffolds Feature Count Comparisons
After annotating Pgenerosa_v074 on 20190701, we noticed a large discrepancy in the number of transcripts that MAKER identified, compared to Pgenerosa_v070. As a reminder, the Pgenerosa_v074 is a subset of Pgenerosa_v070 containing only the top 18 longest scaffolds. So, we decided to do a quick comparison of the annotations present in these 18 scaffolds Pgenerosa_v070 and Pgenerosa_v074.
Genome Annotation - O.lurida 20190709-v081 Transcript Isoform ID with Stringtie on Mox
Earlier today, I generated the necessary Hista2 index, which incorporated splice sites and exons, for use with Stringtie in order to identify transcript isoforms in our 20190709-Olurida_v081 annotation. This annotation utilized tissue-specific transcriptome assemblies provided by Katherine Silliman.
Genome Annotation - O.lurida 20190709-v081 Hisat2 Transcript Isoform Index
Last week I re-annotated our Olurida_v081 genome using tissue-specific transcriptomes. The MAKER annotations don’t yield transcript isoforms, so this is the first part of the process in identifying/annotating different isoforms within the transcriptome.
Genome Assessment - BUSCO Metazoa on Pgenerosa_v074 on Mox
Ran BUSCO on Mox for our Pgenerosa_v74 genome assembly to assess “completeness”. This is the assembly that only has the longest 18 scaffolds (the scaffolds hand-curated by Phase Genomics).
Genome Assessment - BUSCO Metazoa on Pgenerosa_v70 on Mox
Ran BUSCO on Mox for our Pgenerosa_v70 genome assembly to assess “completeness”.
Genome Annotation - Pgenerosa_v071 Using GenSAS
In our various attempts to get the Panopea generosa genome annotated in such a manner that we’re comfortable with (the previous annotation attempts we’re lacking any annotations in almost all of the largest scaffolds, which didn’t seem right), Steven stumbled across GenSAS, a web/GUI-based genome annotation program, so we gave it a shot.
Genome Annotation - Pgenerosa_v074 Using GenSAS
In our various attempts to get the Panopea generosa genome annotated in such a manner that we’re comfortable with (the previous annotation attempts we’re lacking any annotations in almost all of the largest scaffolds, which didn’t seem right), Steven stumbled across GenSAS, a web/GUI-based genome annotation program, so we gave it a shot.
Genome Annotation - Olurida_v081 with MAKER and Tissue-specific Transcriptomes on Mox
I previously annotated our Olurida_v081 genome with MAKER using our “canonical” transcriptome, Olurida_transcriptome_v3.fasta as the EST evidence utilized by MAKER. A discussion on one of our Slack channels related to the lack of isoform annotation (I think it’s a private channel, sorry) prompted Katherine Silliman to suggest re-running the annotation using tissue-specific transcriptome assemblies that she has generated as EST evidence, instead of a singular transcriptome. Since I already had previous versions of the MAKER script that I’ve used for annotations, re-running was rather straightforward. While this was running, I used Stringtie on 20190625to produce a GTF that maps out potential isoforms, as I don’t believe MAKER will actually predict isoforms, since it didn’t do so the first time, nor has it with other annotations we’ve run on geoduck assemblies.
Genome Annotation - Pgenerosa_v074 MAKER on Mox
I previously created a subset of the Pgenerosa_v070 genome assembly that contains just the largest 18 scaffolds (these scaffolds were produced by Phase Genomics, utilizing some Hi-C sequencing). The new subsetted genome is labeled as Pgenerosa_v074.fa (914MB).
Transcriptome Annotation - Geoduck Larvae Day5 EPI99 with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Super Low OA EPI116 with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Juvenile Super Low OA EPI115 with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Juvenile Ambient OA EPI124 with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Juvenile Ambient OA EPI123 with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Gonad with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Ctenidia with Transdecoder on Mox
Used Transdecoder to identify open reading frames (ORFs) for use in annotating Pgenerosa_v074 genome assembly. Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
RepeatModeler - Pgenerosa_v074 for MAKER Annotation on Emu
Yesterday (20190625) I generated a subset of the first 18 FastA sequences from the Pgenerosa_v070.fa file. This subset has been designated as Pgenerosa_v074 by Steven. It’s available on our Genomic Resources wiki:
RepeatMasker - Pgenerosa_v074 for Transposable Element ID on Roadrunner
Yesterday (20190625) I generated a subset of the first 18 FastA sequences from the Pgenerosa_v070.fa file. This subset has been designated as Pgenerosa_v074 by Steven. It’s available on our Genomic Resources wiki:
Data Received - C.virginica Mantle MBD-BSseq from ZymoResearch
We received the BSseq data from ZymoResearch today. Samples were submitted on 20190326. The samples constituted 24 Crassostrea virginica mantle samples which were prepared using the MethylMiner Kit (Invitrogen) on 20190319.
Data Wrangling - FastA Subsetting of Pgenerosa_v070.fa Using samtools faidx
Steven asked to subset the Pgenerosa_v070.fa (2.1GB) in this GitHub Issue #705. In that issue, it was determined that a significant portion of the sequencing data that was assembled by Phase Genomics clustered in “scaffolds” 1 - 18. As such, Steven asked to subset just those 18 scaffolds.
Genome Annotation - O.lurida (v081) Transcript Isoform ID with Stringtie on Mox
Earlier today, I generated the necessary Hista2 index, which incorporated splice sites and exons, for use with Stringtie in order to identify transcript isoforms in our Olurida_v081 annotation.
Genome Annotation - O.lurida (v081) Hisat2 Transcript Isoforms Index
Per this thread in Slack, we realized that the “final” annotation of the Olurida_v081 genome only seemed to have singular mRNA annotations and no apparent isoforms. As such, I decided to see if I could tease out this type of info.
Metagenomics - Refining Anvio Binning
UPDATE 20220121: THIS IS AN INCOMPLETE POST AND IS ONLY POSTED FOR POSTERITY.
Metagenomics - Taxonomic Diversity and Sequencing Coverage with MEGAHIT BLASTx and Krona Plots
After a meeting on this project around the middle of May, we decided to try various approaches to assessing the metagenome. One aspect was to add coverage sequencing coverage information to our BLASTx taxonomy visualizations. I used the MEGAHIT coverage info from 20190327 and the subsequent BLASTx data from 20190516.
Data Wrangling - Create Pgenerosa_v070 GFFs
Since the MAKER genome annotation of our Pgenerosa_v070 genome assembly finally completed last week, I decided to separate out the various features into separate GFF files, as I’m sure we’ll need/want them at some point. This was all done in a Jupyter Notebook on my computer (swoose) - see notebook linked below.
Sample Submission - Tanner Crab Infected vs Uninfected RNAseq
After reviewing our options for sample pooling, we decided to do a comparison of Infected vs. Uninfected crabs.
Metagenomics - BLASTx of Individual Water Sample MEGAHIT Assemblies on Mox
After a meeting on this project yesterda, we decided to try a few things to continue with various approaches to assessing the metagenome. One of the approaches is to run BLASTx on the individual water sample MEGAHIT assemblies from 20190327 and obtain taxonomy info for them, so that’s what I did here.
Library Decisions - C.bairdi RNAs for Library Pools
FastQC-MultiQC - Additional C.gigas WGBS Sequencing Data from Genewiz Received 20190501
Earlier today, we received the additional G.gigas sequencing data from Genewiz. Wanted to run through FastQC again and get an updated report for each data set. Admittedly, it probably won’t look much different from the initial FastQC run on 20190415, due to the fact that the additional sequencing was simply appended to the previous data. Since FastQC examines a subset of the data in each file, I’d fully expect the FastQC report to look the same. However, we’ll have a greater number of sequences in each file. This should, in turn, increase the number of reads retained after quality trimming.
Data Received - Additional C.gigas Whole Genome Bisulfite Sequencing Data from Genewiz
The FastQC analysis of the intitial data we received from Genewiz (on 20190408)showed consistent failures in the “Per Tile Sequence Quality” for all of Roberto’s Crassostrea gigas sequencing. After discussing with Genewiz, they offered to generate an additional 25% reads for each of those libraries.
RNA Isolation and Quantification - C.bairdi Hemolymph Pellet in RNAlater
After the success we had isolating RNA using the Quick-DNA/RNA Microprep Plus Kit (ZymoResearch), Steven had me isolate RNA from a list of ~117 samples. Of that list, I was able to find 66 crab hemolymph pelleted RNAlater samples. The “missing” samples were most likely previosly used by Grace during our various attempts to get some usable RNA out these.
SRA Submission - P.generosa 10x Genomics Sequencing Data
Submitted the 10x Genomics sequencing files to the NCBI short read archive (SRA) that was performed by Illumina, as part of their generous offer to use geoduck samples to test out their new-at-the-time sequencing platform (NovaSeq 6000).
Data Analysis - C.virginica MBD Sequencing Coverage
A while ago, Steven tasked me with assessing some questions related to the sequencing coverage we get doing MBD-BSseq in this GitHub issue. At the heart of it all was really to try to get an idea of how much usable data we actually get when we do an MBD-BSseq project. Yaamini happened to have done an MBD-BSseq project relatively recently, and it’s one she’s actively working on analyzing, so we went with that data set.
Metagenomics Annotation - P.generosa Water Samples Using BLASTn on Mox and KronaTools Visualization to Compare pH Treatments
Nearing the end of this quick metagenomics comparison of taxonomic differences between the two pH treatments (pH=7.1 and pH=8.2). Previously ran:
Metagenomics Gene Prediction - P.generosa Water Samples Using MetaGeneMark on Mox to Compare pH Treatments
Continuing with a relatively quick comparison of pH treatments (pH=7.1 vs. pH=8.2), I wanted to run gene prediction on the MEGAHIT assemblies I made yesterday. I ran MetaGeneMark on the two pH-specific assemblies on Mox. This should be a very fast process (I’m talking, like a couple of minutes fast), so it enhances the annotation with very little effort and time.
FastQC - WGBS Sequencing Data from Genewiz Received 20190408
We received whole genome bisulfite sequencing (WGBS) data from Genewiz last week on 20190408, so ran FastQC on the files on my computer (swoose). FastQC results will be added to Nightingales Google Sheet.
Metagenome Assemblies - P.generosa Water Samples Trimmed HiSeqX Data Using Megahit on Mox to Compare pH Treatments
A report involving our work on the geoduck water metagenomics is due later this week and our in-depth analysis for this project using Anvi’o is likely to require at least another week to complete. Even though we have a broad overview of the metagenomic taxa present in these water samples, we don’t have data in a format for comparing across samples/treatments. So, I initiated our simplified pipeline (MEGAHIT > MetaGeneMark > BLASTn > KronaTools) for examining our metagenomic data of the two treatments:
RNA Isolation and Quantification - Crab Hemolypmh Using Quick-DNA-RNA Microprep Plus Kit
In the continuing struggle to isolate RNA from the Chionoecetes bairdi hemolymph preserved in RNAlater, Pam managed to find the Quick-DNA-RNA Microprep Plus Kit (ZymoResearch) as a potential option. We received a free sample of the kit and so I gave it a shot. Grace pulled 10 samples she’d previously used to isolate RNA (and was unable to get anything out of virtually all of them using the RNeasy Plus Micro Kit (Qiagen)) for me to try out this new kit:
Transcriptome Assembly - Geoduck Tissue-specific Assembly Larvae Day5 EPI99 with HiSeq and NovaSeq Data on Mox
I previously assembled and annotated P.generosa larval Day 5 transcriptome (20190318 - mislabeled as Juvenile Day 5 in my previous notebook entries) using just our HiSeq data from our Illumina collaboration. This was a an oversight, as I didn’t realize that we also had NovaSeq RNAseq data. So, I’ve initiated another de novo assembly using Trinity incorporating both sets of data.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Gonad HiSeq and NovaSeq Data on Mox
I previously assembled and annotated P.generosa gonad transcriptome (20190318) using just our HiSeq data from our Illumina collaboration. This was a an oversight, as I didn’t realize that we also had NovaSeq RNAseq data. So, I’ve initiated another de novo assembly using Trinity incorporating both sets of data.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Juvenile Ambient OA EPI124 with HiSeq and NovaSeq Data on Mox
Ran a de novo assembly on our HiSeq and NovaSeq data from Hollie’s juvenile EPI 124 sample “ambient OA”. This was done for Christian to use in some long, non-coding RNA (lncRNA) analysis.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Juvenile Ambient OA EPI123 with HiSeq Data on Mox
Ran a de novo assembly on our HiSeq data from Hollie’s juvenile EPI 123 sample “ambient OA”. This was done for Christian to use in some long, non-coding RNA (lncRNA) analysis.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Juvenile Super Low OA EPI116 with HiSeq and NovaSeq Data on Mox
Ran a de novo assembly on our HiSeq and NovaSeq data from Hollie’s juvenile EPI 116 sample “super low OA”. This was done for Christian to use in some long, non-coding RNA (lncRNA) analysis.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Juvenile Super Low OA EPI115 with HiSeq Data on Mox
Ran a de novo assembly on our HiSeq data from Hollie’s juvenile EPI 115 sample “super low OA”. This was done for Christian to use in some long, non-coding RNA (lncRNA) analysis.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Ctenidia with HiSeq and NovaSeq Data on Mox
I previously assembled and annotated P.generosa ctenidia transcriptome (20190318) using just our HiSeq data from our Illumina collaboration. This was a an oversight, as I didn’t realize that we also had NovaSeq RNAseq data. So, I’ve initiated another de novo assembly using Trinity incorporating both sets of data.
Data Management - Whole Genome Bisulfite Sequencing Data from Genewiz Received
Received the WGBS data from Genewiz that were sent to Genewiz for whole genome bisulfite sequencing on 20190207. These were from:
Metagenomics - P.generosa Water Sample Assembly Comparisons with Quast
Continuing work on the metagenomics project, Emma shared her “co-assembly”, so I figured it would be quick and easy to compare hers with mine and get a feel for how different/similar they might be. I did a similar comparison last week where I compared each of our individual water sample assemblies. Those results showed my assemblies generated:
Metagenomics - Geoduck Water Sample Assembly Comparisons with MetaQuast
As part of addressing this GitHub Issue on assessing taxonomic diversity in our metagenomics samples, I decided to compare the individual sample assemblies I made on 20190327 using Megahit and the assemblies that Emma made. Emma utilized NGless on her cluster in Genome Sciences with the following scripts:
Metagenomics - Taxonomic Diversity Comparisons from Geoduck Water with Anvio on Mox
I decided to give Anvi’o a shot for this metagenomics analysis, as it seems ridiculously thorough and easy to use, with a nice-looking, interactive plotting interface. Additionally, they have a TON of clearly written tutorials on how to use their software and explanations of what’s happening when you use it! Really, couldn’t ask for much more. So, here’s how it went…
Metagenome Assemblies - P.generosa Water Samples Trimmed HiSeqX Data Using Megahit on Mox
I previously created a single metagenome assembly using all of the sequencing data from this project.
Transposable Element Mapping - Crassostrea gigas Genome v9 Using RepeatMasker 4.07 on Roadrunner
Per this GitHub issue, I’m IDing transposable elements (TEs) in the Crassostrea gigas genome. Even though the C.gigas genome should be fully annotated, Steven wants a comparable set of analyses to compare to the Crassostrea virginica TE mapping we previously performed.
Sample Submission - Lotterhos C.virginica Mantle MBD DNA to ZymoResearch for BSseq
Crassostrea virginica samples that were subjected to MBD enrichment (completed 20190319) were shipped, on dry ice, to ZymoResearch today for BSseq. They will perform bisulfite conversion, library prep, and sequencing. Sequencing output is to be ~50M paired-end reads (at least 100bp or 151bp) per library.
Data Management - SRA Submission Panopea generosa Day 5 Larvae RNAseq
In preparation for some manuscripts, Steven asked that I get some geoduck RNAseq data upload to the NCBI sequencing read archive (SRA) in this GitHub Issue. Specifically, he needed the data corresponding to day 5 (pos-fertilization) larve RNAseq data that was prepped/run by Illumina on the NovaSeq. Original sample submission notebook entry is here.
Metagenomics - Taxonomic Diversity from Geoduck Water with BLASTn and Krona Plots
Continuing on getting the metagenomics sequencing project written up as a manuscript, Steven asked me to provide an overview of the taxonomic makeup of our metagenome assembly in this GitHub Issue. Earlier today, I ran analysis using BLASTp data. I initiated additional analysis using the MetaGeneMark nucleotide data to run BLASTn on Mox.
Metagenomics - Taxonomic Diversity from Geoduck Water with BLASTp and Krona plots
We’re working on getting the metagenomics sequencing project written up as a manuscript and Steven asked me to provide an overview of the taxonomic makeup of our metagenome assembly in this GitHub Issue.
MBD Enrichment - DNA Quantification of C.virginica MBD Samples from 20190312
Finished MBD enrichment on 20190312 using the MethylMiner kit. Since we were out of Qubit assay tubes, I could not quantify these samples when I initially completed the ethanol precipitation. Tubes are in, so I went forward with quantification using the Roberts’ Lab Qubit 3.0 and the 1x High Sensitivity dsDNA assay.
Transcriptome Annotation - Geoduck Juvenile Day 5 with Trinotate on Mox
Transcriptome Annotation - Geoduck Heart with Trinotate on Mox
Transcriptome Annotation - Geoduck Gonad with Trinotate on Mox
Transcriptome Annotation - Geoduck Ctenidia with Trinotate on Mox
Transcriptome Annotation - Geoduck Heart with BLASTx on Mox
I’ll be annotating the transcriptome assembly (from 20190215) using Trinotate and part of that process is having BLASTx output for the Trinity assembly, so have run BLASTx on Mox.
Transcriptome Annotation - Geoduck Gonad with BLASTx on Mox
I’ll be annotating the transcriptome assembly (from 20190215) using Trinotate and part of that process is having BLASTx output for the Trinity assembly, so have run BLASTx on Mox.
Transcriptome Annotation - Geoduck Ctenidia with BLASTx on Mox
I’ll be annotating the transcriptome assembly (from 20190215) using Trinotate and part of that process is having BLASTx output for the Trinity assembly, so have run BLASTx on Mox.
Transcriptome Annotation - Geoduck Juvenile Day 5 with BLASTx on Mox
I’ll be annotating the transcriptome assembly (from 20190215) using Trinotate and part of that process is having BLASTx output for the Trinity assembly, so have run BLASTx on Mox.
Transcriptome Annotation - Geoduck Juvenile Day 5 with Transdecoder on Mox
Continuing tissue-specific RNAseq. Using Transdecoder to identify open reading frames (ORFs). Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Heart with Transdecoder on Mox
Continuing tissue-specific RNAseq. Using Transdecoder to identify open reading frames (ORFs). Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Gonad with Transdecoder on Mox
Continuing tissue-specific RNAseq. Using Transdecoder to identify open reading frames (ORFs). Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Transcriptome Annotation - Geoduck Ctenidia with Transdecoder on Mox
Continuing tissue-specific RNAseq. Using Transdecoder to identify open reading frames (ORFs). Relies on BLASTp, Pfam, and HMM scanning to ID ORFs.
Methylation Analysis - C.virginica Gonad MBD with Varying Read Subsets with Bismark on Mox
Steven asked for the following analysis in this GitHub Issue using Yaamini’s C.virginica MBD samples:
Data Management - Create C.virginica Bisulfite Genome with Bismark on Mox
In preparation for some a bisulfite analysis requested by Steven, I needed to make bisulfite genome with, and for, Bismark to use and wanted to use the same one Yaamini has been working with. Downloaded the NCBI version of the C.virginica genome.
MBD Enrichment - Ethanol Precipitation of C.virginica MBD samples
After completing two rounds of MBD enrichment (first 12 samples on 20190307 and the second 12 samples on 20190311), I performed ethanol precipitation, per the MethylMiner instructions with the following modifications:
MBD Enrichment - C.virginica Sheared Mantle DNA from 20190306
Last week I performed MBD enrichment on 12 of the 24 Lotterhos C.viriginica mantle DNA on samples I had sheared on 20190306. I proceeded with MBD enrichment using the MethylMiner Kit (Invitrogen) on the 12 remaining samples of the 24. I followed the manufacturer’s protocol for input DNA >1ug - 10ug with the following modifications:
MBD Enrichment - C.virginica Sheared Mantle DNA from 20190306
Yesterday, I sheared the Lotterhos C.virginica gDNA in preparation for MBD enrichment. Today, I proceeded with MBD enrichment using the MethylMiner Kit (Invitrogen) on 12 of the 24 samples. I followed the manufacturer’s protocol for input DNA >1ug - 10ug with the following modifications:
DNA Shearing & Bioanalyzer - Lotterhos C.virginica Mantle gDNA from 2018114
Crassostrea virginica mantle gDNA that had previously been isolated on 20181114 was evaluated for integrity via agaroase gel yesterday (20190305). Everything looked pretty good, so proceeded with shearing in preparation for MBD enrichment.
Agarose Gel - Lotterhos C.virginica Mantle DNA from 20181114
Checked DNA integrity of the Crassostrea virginica mantle gDNA I isolated on 20181114 in preparation for MBD enrichment. Detailed sample info and sample processing (including calculations for MBD enrichment using the MethylMiner Kit) are here (Google Sheet):
Data Management - Data Migration and Drive Expansion on Gannet
A little while ago, we installed some additional hard drives in Gannet (Synology RS3618XS) with the intention of expanding the total storage space. However, the original set of HDDs were set up as RAID10. As it turns out, RAID10 configurations cannot be expanded! So, the new set of HDDs were configured as a separate volume (Volume 2) in a RAID6 configuration. After backing up the /volume1/web directory (via rsync) to our UW Google Drive, I begane the data migration.
Genome Assessment - BUSCO Metazoa on P.generosa v071 on Mox
Ran BUSCO on our completed annotation of the P.generosa v071 genome (GFF) (subset of sequences >10kbp). See this notebook entry for genome annotation info. This provides a nice metric on how “complete” a genome assembly (or transcriptome) is. Additionally, BUSCO is tied in with Augustus for gene prediction and generates ab initio gene models. With that said, since I just want to evaluate the completeness of this particular genome assembly, I’ll be using the annotated genome generated through two rounds of SNAP gene prediction. Otherwise, I’d use the initial MAKER annotations to generate an Augustus gene model that could be used in conjuction with the SNAP models (I’ll likely do this at a later date).
Genome Annotation - Pgenerosa_v070 MAKER on Mox
Here it goes, a massive undertaking of attempting to annotate the Pgenerosa_v070 genome (FastA; 2.1GB) using MAKER on Mox! I previously started this on 20190115, but killed it in order to fix a number of different issues with the script that were causing problems. I decided the changes were substantial enough, that I’d just make a new working directory and notebook entry.
Data Wrangling - CpG OE Calculations on C.virginica Genes
Steven tasked me with processing ~90 FastA files containing gene sequences from C.virginica in this GitHub Issue. He needed to determine the Observed/Expected (O/E) ratio of CpGs in each FastA. He provided this example code and this link to all the files. Additionally, today, he tasked Kaitlyn with merging all of the output CpG O/E values for each sample in to a single file, but I decided to tackle it anyway.
Methylation Analysis - P.generosa Bismark on Mox
This is a quick and dirty (i.e. no adaptor/quality trimming) assessment of all of our Panopea generosa bisulfite sequencing efforts to date in order to get a rough idea of methylation mapping, per this GitHub issue. Ran Bismark on Mox on a series of subset of the reads:
Methylation Analysis - O.lurida Bismark on Mox
This is a quick and dirty (i.e. no adaptor/quality trimming) assessment of all of our Ostrea lurida bisulfite sequencing efforts to date in order to get a rough idea of the methylation mapping, per this GitHub issue. Ran Bismark on Mox on a series of subset of the reads:
Methylation Analysis - C.virginica
This is a quick and dirty (i.e. no adaptor/quality trimming) assessment of all of our Crassostrea irginica bisulfite sequencing efforts to date in order to get a rough idea of the methylation mapping, per this GitHub issue. Ran Bismark on Mox on a series of subset of the reads:
Methylation Analysis - C.gigas Bismark on Mox
This is a quick and dirty (i.e. no adaptor/quality trimming) assessment of all of our Crassostrea gigas bisulfite sequencing efforts to date in order to get a rough idea of the methylation mapping, per this GitHub issue. Ran Bismark on Mox on a series of subset of the reads:
Data Management - Create C.virginica Bisulfite Genome wit Bismark on Mox
In preparation for some a quick and “dirty” bisulfite analysis, I needed to make bisulfite genomes with, and for, Bismark to use.
Data Management - Create C.gigas Bisulfite Genome with Bismark on Mox
In preparation for some a quick and “dirty” bisulfite analysis, I needed to make bisulfite genomes with, and for, Bismark to use.
Data Management - Create Geoduck Bisulfite Genomes with Bismark on Mox
In preparation for some a quick and “dirty” bisulfite analysis, I needed to make bisulfite genomes with, and for, Bismark to use.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Juvenile Day 5
Based on a discussion in this GitHub Issue, I’ve initiated some tissue-specific transcriptome assemblies with our current geoduck data.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Heart
Based on a discussion in this GitHub Issue, I’ve initiated some tissue-specific transcriptome assemblies with our current geoduck data.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Gonad
Based on a discussion in this GitHub Issue, I’ve initiated some tissue-specific transcriptome assemblies with our current geoduck data.
Transcriptome Assembly - Geoduck Tissue-specific Assembly Ctenidia
Based on a discussion in this GitHub Issue, I’ve initiated some tissue-specific transcriptome assemblies with our current geoduck data.
Genome Annotation - Pgenerosa_v71 with MAKER on Mox
With the P.generosa v070 annotation taking a very long time (due to the genome/transcriptome sizes, and, hitting some snags in the script I put together), Steven asked if I could subset the genome and annotate the v071 assembly (>10kbp subset) in order to have some annotations to use for some talks coming up ASLO (in this GitHub issue).
Sequence Subsetting - Pgenerosa_v70 Genome Assembly with faidx
Steven asked to subset geoduck assembly in to >10kbp and >30kbp groups. Here are the commands, using faidx:
Samples Submitted - Robertos C.gigas DNA for Whole Genome Bisulfite Sequencing (Genewiz)
Sent Roberto’s 12 designated DNA samples for whole genome bisulfite sequencing (WGBS) to Genewiz. They will do all bisulfite conversion, library prep and sequencing (Illumina HiSeq; 2x150bp; 30x coverage).
Samples Received - C.virginica DNA and Tissues from Lotterhos Lab
Sample list is here (Google Sheet):
DNA Isolation and Quantification - Ronit’s C.gigas Ploidy Ctenidia
Last week, I isolated DNA from all of Ronti’s ctenidia samples, however one sample (D18-C) didn’t isolate properly. So, I performed another isolation procedure with another section of frozen tissue. Tissue was excised from frozen tissue block via razor blade (weight not recorded) and pulverized under liquid nitrogen. Samples were incubated O/N @ 37oC (heating block) in 350uL of MB1 Buffer + 25uL Proteinase K, per the E.Z.N.A. Mollusc DNA Kit (Omega) instructions.
DNA Isolation - C.gigas Ploidy Experiment Ctenidia
Isolated DNA from the remaining ctenidia tissue samples from Ronit’s experiment (Google Sheet).
Annotation - Olurida_v081 MAKER BUSCO Metazoa Augustus Training on Mox
Previously performed this analysis using the eukaryota_odb9 BUSCO database. Figured I’d give it another go using a more specific database, metazoa_odb9, for comparison.
Annotation - Geoduck Genome with MAKER Submitted to Mox
Well, here we go! Initiating full-blown annotation of the Pgenerosa_v070 (FastA; 2.1GB), using MAKER (v.2.31.10) on Mox. This will perform the following:
Annotation - Olurida_v081 MAKER BUSCO (eukaryota_odb9) Augustus Training Submitted to Mox
I’ve previously run MAKER, followed by two round of SNAP gene predictions and now I’m going to throw a BUSCO/Augustus gene training & prediction in the mix to see how it compares. In order to do so, I’ve followed this GitHub Gist on using MAKER/SNAP/BUSCO/Augustus as a guide on what the process should entail.
Annotation - Olurida_v081 MAKER Functional Annotations on Mox
Believe it or not, full, proper annotation of the Olympia oyster genome is nearly complete! Here’s where it stands:
Annotation - Olurida_v081 MAKER Proteins BLASTp on Mox
After getting through the initial MAKER annotation and SNAP gene predictions, and then renaming the sequences, I needed to run BLASTp on the FastA file produced by MAKER id mapping in order to assign functionality to the predicted genes.
Annotation - Olurida_v081 MAKER Proteins InterProScan5 on Mox
Continuation of genome annotation of the Olympia oyster genome. Determined initial gene models using MAKER with two rounds of SNAP, relabeled with more user-friendly names, and then performed protein-level annotations using BLASTp. Next, I’m going to run InterProScan5 (IPS5) to help functionally characterize the O.lurida proteins ID’d by MAKER. Once this is complete, I’ll use MAKER to incorporate the IPS5 and BLASTp results into a much more neatly (i.e. human-readable) annotated genome!
Annotation - Olurida_v081 MAKER ID Mapping
After getting through the initial MAKER annotation and SNAP gene predictions, I needed (wanted?) to simplify annotation names that will be easier to read and are in a more standardized format; similiar to NCBI.
Gene Prediction - HiSeqX Metagenomics from Geoduck Water Using MetaGeneMark on Mox
After assembline the metagenomic data yesterday, I needed to predict some genes. I did this using MetaGeneMark (v.3.38) and ran it on Mox.
Metagenome Assembly - P.generosa Water Sample Trimmed HiSeqX Data Using Megahit on Mox
Attempting an assembly of our geoduck metagenomics HiSeqX data using Megahit (v.1.1.4) on a Mox node.
VCF Splitting - C.virginica VCF Using BCFtools
2018
DNA Isolation - C.gigas Ploidy Experiment Ctenidia
Yesterday, Ronit initiated DNA isolation from ctenidia samples from his experiment (Google Sheet) from the following four samples:
Repeat Library Construction - P.generosa RepeatModeler v1.0.11
Needed to generate a generate a species-specific repeat library for use with MAKER genome annotation using RepeatModeler and the Pgenerosa_v070.fa (see our Genomic Resources wiki for more info) version of the genome.
qPCR - Relative mitochondrial abundance in C.gigas diploids and triploids subjected to acute heat stress via COX1
Using the C.gigas cytochrome c oxidase (COX1) primers (SR IDs: 1713, 1714)I designed the other day, I ran a qPCR on a subset of Ronit’s diploid/triploid control/heat shocked oyster DNA that Shelly had previously isolated and performed global DNA methylation assay. The goal is to get a rough assessment of whether or not there appear to be differences in relative mitochondrial abundances between these samples.
BLASTx - Clupea pallasii (Pacific herring) liver and testes transcriptomes on Mox
Apparently we’ve had some interest in the Pacific herring transcriptomes (liver and testes) we produced many years ago. As such, Steven asked that I do a quick BLASTx to help annotate these two transcriptomes.
FastQC and Trimming - Metagenomics (Geoduck) HiSeqX Reads from 20180809
Steven tasked me with assembling our geoduck metagenomics HiSeqX data. The first part of the process is examining the quality of the sequencing reads, performing quality trimming, and then checking the quality of the trimmed reads. It’s also possible (likely) that I’ll need to run another round of trimming. The process is documented in the Jupyter Notebook linked below. After these reads are cleaned up, I’ll transfer them over to our HPC nodes (Mox) and try assembling them.
Primer Design - Gigas COX1 using Primer3
We’re attempting a quick & dirty comparison of relative mitochondria counts between diploid and triploid C.gigas, so needed a single-copy mitochondrial gene target for qPCR. Selected cytochrome c oxidase subunit 1 (COX1), based on a quick glance at the NCBI mt genome browser for C.gigas NC_001276.
qPCR - Geoduck gonad cDNA with vitellogenin primers
Earlier today I made some cDNA from geoduck gonad RNA for use in this qPCR to test out the vitellogenin primers I designed on 20181129
Reverse Transcription - Geoduck gonad RNA pool
To be able to actually test the vitellegenin primers I made, I needed some geoduck cDNA.
Primer Design - Geoduck Vitellogenin using Primer3
In preparation for designing primers for developing a geoduck vitellogenin qPCR assay, I annotated a de novo geoduck transcriptome assembly last week. Next up, identify vitellogenin genes, design primers, confirm their specificity, and order them!
Annotation - Olurida_v081 MAKER on Mox
Remarkably, I managed to burn through our Xsede computing resources and don’t have terribly much to show for it. Ooof! This is a major bummer, as it “only” takes ~8hrs for a WQ-MAKER job to run there, as opposed to months the last time I tried running it on Mox. Although we have used up our Xsede allocation, all is not lost! The experience of setting up/running WQ-MAKER has enlightened me on how it all works and how to run it on Mox so it will (hopefully) take far, far less time than the last Mox attempt. With that said, here we go…
Annotation - Geoduck Transcritpome with TransDecoder
I was tasked with generating some qPCR primers to analyze vitellogenin expression in geoduck. In order to do so, I needed to annotate a geoduck transcriptome in order to identify potential vitellogenin genes. I had previously assembled a geoduck transcriptome. For annotation, I used TransDecoder (v5.5.0). The annotation was run on our Mox HPC node.
Reverse Transcription - Ronit’s C.gigas DNased RNA from 20181115
Quantified Ronit’s DNased RNA earlier today and proceeded with reverse transcription using 100ng of DNased RNA with oligo dT primers (Promega) and M-MLV reverse transcriptase (Promega) according to the manufacturer’s protocol.
RNA Quantification - Ronit’s C.gigas DNased RNA from 20181115
After confirming Ronit’s DNased RNA was free of gDNA, I quantified the DNased RNA from 20181115 using the Roberts Lab Qubit 3.0 and the Qubit hsRNA Assay.
qPCR - Ronit’s DNased C.gigas RNA with Elongation Factor Primers
After DNasing Ronit’s RNA earlier today, I needed to verify the RNA was free of any contaminating gDNA.
DNase - Ronit’s C.gigas Ctenidia RNA
DNA Isolation and Quantification - Lotterhos C.virginica Mantle DNA
Isolated DNA from the Lotterhos Crassostrea virginica mantle samples received on 20181017 using the E.Z.N.A. Mollusc Kit, with the following modifications/notes:
Computing - Install NCBI nr nt BLAST Database on Mox
Per this issue on GitHub, I installed the pre-formatted NCBI non-redudant (nr) nucleotide (nt) database on Mox.
My New Notebook
Here we are - my new notebook!
Repeat Library Construction - O.lurida RepeatModeler v1.0.11
In further attempts to improve our Ostrea lurida genome annotation, I decided to generate a species-specific repeat library for use with MAKER genome annotation using RepeatModeler.
qPCRs - Ronit’s C.gigas ploidy/dessication/heat stress cDNA (1:5 dilution)
IMPORTANT: The cDNA used for the qPCRs described below was a 1:5 dilution of Ronit’s cDNA made 20181017 with the following primers! Diluted cDNA was stored in his -20oC box with his original cDNA.
Samples Received - Crassostrea virginica (Eastern oyster) tissue from Lotterhos Lab (Northeastern University)

Reverse Transcription - Ronit’s C.gigas DNased ctenidia RNA
Proceeded with reverse transcription of [Ronit’s DNased ctenidia RNA (from 20181016)(2018-10-16-dnase-treatment-ronits-c-gigas-ploiyddessication-ctenidia-rna.html).
qPCR - Ronit’s DNAsed C.gigas Ploidy/Dessication RNA with elongation factor primers
After I figured out the appropriate DNA and primers to use to detect gDNA in Crassostrea gigas samples, I checked Ronit’s [DNased ctenidia RNA (from 20181016)(2018-10-16-dnase-treatment-ronits-c-gigas-ploiyddessication-ctenidia-rna.html) for residual gDNA.
qPCR – C.gigas primer and gDNA tests with 18s and EF1 primers
The qPCR I ran earlier today to check for residual gDNA in Ronit’s DNased RNA turned out terribly, due to a combination of bad primers and, possibly, bad gDNA.
qPCR - Ronit’s DNAsed C.gigas Ploidy/Dessication RNA with 18s primers
After DNasing Ronit’s RNA earlier today, I needed to check for any residual gDNA.
DNase Treatment - Ronit’s C.gigas Ploiyd/Dessication Ctenidia RNA
After quantifying Ronit’s RNA earlier today, I DNased them using the Turbo DNA-free Kit (Ambion), according to the manufacturer’s standard protocol.
RNA Quantification - Ronit’s C.gigas Ploidy/Dessication RNA
Last Friday, Ronit quantified 1:10 dilutions of the RNA I isolated on 20181003 and the RNA he finished isolating on 20181011, but two of the samples (D11-C, T10-C) were still too concentrated.
Data Received - Chionoecetes bairdi RNAseq & FastQC Analysis
We received Grace’s 100bp PE NovaSeq (Illumian) RNAseq data from the Northwest Genomics Center today.
VCF Splitting with bcftools
RNA Isolation - Tanner Crab Hemolymph Pellet in RNAlater using TriReagent
I previously isolated RNA from crab hemolymp from a lyophilized sample using TriReagent and Grace recently tried isolating RNA from crab hemolyph pellet (non-lyophilized) using TriReagent. The results for her extractions weren’t so great, so I’m giving it a shot with the following samples:
RNA Isolation - Ronit’s C.gigas diploid/triploid dessication/heat shock ctenidia tissues
Isolated RNA from a subset of Ronit’s Crassostrea gigas ctenidia samples (see Ronit’s notebook for experiment deets):
SRA Submission - Olymia oyster Whole Genome BS-seq Data
Submitted our whole genome bisulfite sequencing data to NCBI Sequence Read Archive (SRA).
Installation - Microsoft Machine Learning Server (Microsoft R Open) on Emu/Roadrunner R Studio Server
Steven recently saw an announcement that Microsoft R Open now handles multi-threaded processing (default R does not), so we were interested in trying it out. I installed MLR/MRO on Emu/Roadrunner (Apple Xserve; Ubuntu 16.04). Followed the Microsoft installation directions for Ubuntu. In retrospect, I think I could’ve just installed MRO, but this gets the job done as well and won’t hurt anything.
Ubuntu – Fix “No Video Signal” Issue on Emu
An issue with Emu cropped up a few weeks ago that was seemingly caused by upgrading from Ubuntu 16.04 to 18.04.
Transcriptome Alignment & Bedgraph – Olympia oyster transcriptome with Olurida_v080 genome assembly
Bedgraph – Olympia oyster transcriptome with Olurida_v081 genome assembly
I took the sorted BAM file from yesterday’s corrected RNAseq genome alignment and converted it to a bedgraph using BEDTools genomeCoverageBed tool.
Transcriptome Alignment – Olympia oyster RNAseq reads aligned to genome with HISAT2
Yesterday’s attempt at producing a bedgraph was a failure and a prodcuct of a major brain fart. The worst part is that I was questioning what I was doing the entire time, but still went through with the process! Yeesh!
Bedgraph - Olympia oyster transcriptome (FAIL)
Progress on generating bedgraphs from our Olympia oyster transcriptome continues.
Transcriptome Alignment - Olympia oyster Trinity transcriptome aligned to genome with Bowtie2
Progress on generating bedgraphs from our Olympia oyster transcriptome continues.
Transcriptome Assembly - Olympia oyster RNAseq Data with Trinity
Used all of our current oly RNAseq data to assemble a transcriptome using Trinity.
DNA Methylation Analysis – Olympia Oyster Whole Genome BSseq Bismark Pipeline MethylKit Comparison
I previously ran two variations on the Bismark analysis for our Olympia oyster whole genome bisulfite sequencing data:
RNA Isolation - Lyophilized Tanner Crab Hemolymph in RNAlater
Due to difficulties getting RNA from hemolymph samples stored in RNAlater, Grace is testing out lyophilizing samples before extraction. Who knows what impact this will have on RNA, but it’s worth a shot!
DNA Methylation Analysis - Olympia Oyster Whole Genome BSseq Bismark Pipeline Comparison
Ran Bismark using our high performance computing (HPC) node, Mox, with two different bowtie2 settings:
DNA Quantification - Sea Lice DNA from 20180523
We previously received sea lice (Caligus tape) DNA from Cris Gallardo-Escarate at Universidad de Concepción.
TrimGalore/FastQC/MultiQC - C.virginica Oil Spill MBDseq Concatenated Sequences
Sequencing Data Analysis - C.virginica Oil Spill MBDseq Concatenation & FastQC
Per Steven’s request, I concatenated our Crassostrea virginica LSU oil spill MBDseq sequencing data and ran FastQC on the concatenated files.
DNA Methylation Analysis - Olympia oyster BSseq MethylKit Analysis
NOTE: IMPORTANT CAVEATS - READ POST BEFORE USING DATA.
Transcriptome Assembly - Geoduck RNAseq data
Used all of our current geoduck RNAseq data to assemble a transcriptome using Trinity.
FastQC/MultiQC/TrimGalore/MultiQC/FastQC/MultiQC - O.lurida WGBSseq for Methylation Analysis
I previously ran this data through the Bismark pipeline and followed up with MethylKit analysis. MethylKit analysis revealed an extremely low number of differentially methylated loci (DML), which seemed odd.
Transposable Element Mapping – Crassostrea virginica Genome, Cvirginica_v300, using RepeatMasker 4.07
Per this GitHub issue, I’m IDing transposable elements (TEs) in the Crassostrea virginica genome.
Assembly Stats – Geoduck Hi-C Final Assembly Comparison
We received the final geoduck genome assembly data from Phase Genomics, in which they updated the assembly by performing some manual curation:
DNA Methylation Analysis - Bismark Pipeline on All Olympia oyster BSseq Datasets
Bismark analysis of all of our current Olympia oyster (Ostrea lurida) DNA methylation high-throughput sequencing data.
Data Received - Geoduck Metagenome HiSeqX Data
Received the data from the geoduck metagenome libraries that I prepared and were sequenced at the Northwest Genomics Center at UW on the HiSeqX (Illumina) - PE 151bp.
RNA Isolation & Quantificaiton - Tanner Crab Hemolymph
Isolated RNA from 40 Tanner crab hemolymph samples selected by Grace with the RNeasy Plus Micro Kit (Qiagen) according to the manufacturer’s protocol, with the following modifications:
Genome Annotation – Olympia oyster genome annotation results #02
Genome Annotation - Olympia oyster genome annotation results #01
Genome Annotation – Olympia oyster genome complete - brief note
Whoa! Genome annotation using Jetstream/WQ-MAKER that I started this morning is complete!! Only 7hrs!
Genome Annotation - Olympia oyster genome using WQ-MAKER Instance on Jetstream
Yesterday, our Xsede Startup Application (Google Doc) got approval for 100,000 Service Units (SUs) and 1TB of disk space on Xsede/Atmosphere/Jetstream (or, whatever it’s actually called!). The approval happened within an hour of submitting the application!
Bioanalyzer - Tanner Crab RNA Isolated with RNeasy Plus Mini Kit
Ran the four Tanner crab RNA samples that I isolated yesterday on the Seeb Lab Bioanalyzer 2100 (Agilent) using the RNA Pico 6000 Kit.
RNA Cleanup - Tanner Crab RNA
In a continued attempt to figure out what we can do about the tanner crab RNA, Steven tasked me with using an RNeasy Kit to cleanup some existing RNA.
RNA Isolation - Tanner Crab Hemolymph Using RNeasy Plus Mini Kit
Tanner crab RNA has proved a bit troublesome. As such, [Steven asked me to try isolating some RNA using the RNeasy Plus Mini Kit (Qiagen)(https://github.com/RobertsLab/resources/issues/327) to see how things would turn out.
Mox – Over quota: Olympia oyster genome annotation progress (using Maker 2.31.10)
UPDATE 20180730
RNA Cleanup - Tanner Crab RNA Pools
Grace had previously pooled a set of crab RNA in preparation for RNAseq. Yesterday, we/she concentrated the samples and then quantified them. Unfortunately, Qubit results were not good (concentrations were far below the expected 20ng/uL) and the NanoDrop1000 results yielded awful looking curves.
Mox - Olympia oyster genome annotation progress (using Maker 2.31.10)
TL;DR - It appears to be continuing where it left off!
Mox - Password-less SSH!
The high performance computing (HPC) cluster (called Mox) at Univ. of Washington (UW) frustratingly requires a password when SSH-ing, even when SSH keys are in use. I have a lengthy, unintelligible password that I use for my UW account, so having to type this in any time I want to initiate a new SSH session on Mox is a painful process.
Ubuntu - Fix “No Video Signal” Issue on Emu/Roadrunner
Both Apple Xserves (Emu/Roadrunner) running Ubuntu (16.04LTS) experienced the same issue - the monitor would indicate “No Video Signal”, would go dark, and wasn’t responsive to keyboard/mouse movements. However, you could ssh into both machines w/o issue.
Transposable Element Mapping – Olympia Oyster Genome Assembly, Olurida_v081, using RepeatMasker 4.07
I previously performed this analysis using a different version of our Ostrea lurida genome assembly. Steven asked that I repeat the analysis with a modified version of the genome assembly (Olurida_v081) - only has contigs >1000bp in length.
Library Construction - Geoduck Water Filter Metagenome with Nextera DNA Flex Kit (Illumina)
Made Illumina libraries with goeduck metagenome water filter DNA I previously isolated on:
BS-seq Mapping – Olympia oyster bisulfite sequencing: Bismark Continued
Transposable Element Mapping – Crassostrea virginica NCBI Genome Assembly using RepeatMasker 4.07
Genome used: NCBI GCA_002022765.4_C_virginica-3.0
Read Mapping – Olympia oyster 2bRAD Data with Bowtie2 (on Mox)
Transposable Element Mapping - Olympia Oyster Genome Assembly using RepeatMasker 4.07
DNA Received - Sea lice DNA from Cris Gallardo-Escarate at Universidad de Concepción
Received Caligus tape DNA - two samples:
Software Installation – RepeatMasker v4.0.7 on Emu/Roadrunner Continued
After yesterday’s difficulties getting RMblast to compile, I deleted the folder and went through the build process again.
Software Installation - RepeatMasker v4.0.7 on Emu/Roadrunner
Steven asked that I re-run some Olympia oyster transposable elements analysis using RepeatMasker and a newer version of our Olympia oyster genome assembly.
TrimGalore/FastQC/MultiQC – TrimGalore! RRBS Geoduck BS-seq FASTQ data (directional)
Earlier this week, I ran TrimGalore!, but set the trimming, incorrectly - due to a copy/paste mistake, as --non-directional, so I re-ran with the correct settings.
FastQC - RRBS Geoduck BS-seq FASTQ data
Earlier today I finished trimming Hollie’s RRBS BS-seq FastQ data.
TrimGalore/FastQC/MultiQC – TrimGalore! RRBS Geoduck BS-seq FASTQ data
Data Management - Illumina NovaSeq Geoduck Genome Sequencing
As part of the Illumina collaborative geoduck genome sequencing project, their end goal has always been to sequence the genome in a single run.
Assembly - Geoduck Hi-C Assembly Subsetting
Read Mapping - Mapping Illumina Data to Geoduck Genome Assemblies with Bowtie2
We have an upcoming meeting with Illumina to discuss how the geoduck genome project is coming along and to decide how we want to proceed.
BS-seq Mapping - Olympia oyster bisulfite sequencing: TrimGalore > FastQC > Bismark
Assembly & Stats - SparseAssembler (k95) on Geoduck Sequence Data > Quast for Stats
Had a successful assembly with SparseAssembler k101, but figured I’d just tweak the kmer setting and throw it in the queue and see how it compares; minimal effort/time needed.
Assembly Stats - Geoduck Genome Assembly Comparisons w/Quast - SparseAssembler, SuperNova, Hi-C
Assembly Stats - Geoduck Hi-C Assembly Comparison
Ran the following Quast command to compare the two geoduck assemblies provided to us by Phase Genomics:
DNA Isolation & Quantification - Metagenomics Water Filters
After discussing the preliminary DNA isolation attemp with Steven & Emma, we decided to proceed with DNA isolations on the remaining 0.22μm filters.
Total Alkalinity Calculations - Yaamini’s Ocean Chemistry Samples
I ran a subset of Yaamini’s ocean chemistry samples on our T5 Excellence titrator (Mettler Toledo) at the beginning of April. The subset were samples taken from the beginning, middle, and end of the experiment. The rationale for this was to assess whether or not total alkalinity (TA) varied across the experiment. If there was little variation, then there’d likely be no need to run all of the samples. However, should there be temporal differences, then all samples should be processed.
Assembly – SparseAssembler (k 111) on Geoduck Sequence Data
Continuing to try to find the best kmer setting to work with SparseAssemlber after the last attempt failed due to a kmer size that was too large (k 131; which happens to be outside the max kmer size [127] for SparseAssembler), I re-ran SparseAssembler with an arbitrarily selected kmer size < 131 (picked k 111).
Assembly – SparseAssembler (k 131) on Geoduck Sequence Data
After some runs with kmergenie, I’ve decided to try re-running SparseAssembler using a kmer setting of 131.
Data Management - Geoduck Phase Genomics Hi-C Data
We received sequencing/assembly data from Phase Genomics.
Kmer Estimation – Kmergenie (k 301) on Geoduck Sequence Data
Continuing the quest for the ideal kmer size to use for our geoduck assembly.
Kmer Estimation – Kmergenie Tweaks on Geoduck Sequence Data
Earlier today, I ran kmergenie on our all of geoduck DNA sequencing data to see what it would spit out for an ideal kmer setting, which I would then use in another assembly attempt using SparseAssembler; just to see how the assembly might change.
Kmer Estimation - Kmergenie on Geoduck Sequence Data (default settings)
After the last SparseAssembler assembly completed, I wanted to do another run with a different kmer size (last time was arbitrarily set at 101). However, I didn’t really know how to decide, particularly since this assembly consisted of mixed read lenghts (50bp and 100bp). So, I ran kmergenie on all of our geoduck (Panopea generosa) sequencing data in hopes of getting a kmer determination to apply to my next assembly.
Assembly Stats - Quast Stats for Geoduck SparseAssembler Job from 20180405
The geoduck genome assembly started 20180405 completed this weekend.
Data Management - SRA Submission LSU C.virginica Oil Spill MBD BS-seq Data
Submitted the Crassostrea virginica (Eastern oyster) MBD BS-seq data we received on 20150413 to NCBI Sequence Read Archive.
TrimGalore/FastQC/MultiQC - Trim 10bp 5’/3’ ends C.virginica MBD BS-seq FASTQ data
Steven found out that the Bismarck documentation (Bismarck is the bisulfite aligner we use in our BS-seq pipeline) suggests trimming 10bp from both the 5’ and 3’ ends. Since this is the next step in our pipeline, we figured we should probably just follow their recommendations!
DNA Isolation & Quantification - Metagenomics Water Filters
Isolated DNA from the following two filters:
TrimGalore/FastQC/MultiQC - 2bp 3’ end Read 1s Trim C.virginica MBD BS-seq FASTQ data
Earlier today, I ran TrimGalore/FastQC/MultiQC on the Crassostrea virginica MBD BS-seq data from ZymoResearch and hard trimmed the first 14bp from each read. Things looked better at the 5’ end, but the 3’ end of each of the READ1 seqs showed a wonky 2bp blip, so decided to trim that off.
TrimGalore/FastQC/MultiQC - 14bp Trim C.virginica MBD BS-seq FASTQ data
Yesterday, I ran TrimGalore/FastQC/MultiQC on the Crassostrea virginica MBD BS-seq data from ZymoResearch with the default settings (i.e. “auto-trim”). There was still some variability in the first ~15bp of the reads and Steven wanted to see how a hard trim would change things.
TrimGalore/FastQC/MultiQC - Auto-trim C.virginica MBD BS-seq FASTQ data
Yesterday, I ran FastQC/MultiQC on the Crassostrea virginica MBD BS-seq data from ZymoResearch. Steven wanted to trim it and see how things turned out.
FastQC/MultiQC - C. virginica MBD BS-seq Data
Per Steven’s GitHub Issues request, I ran FastQC on the Eastern oyster MBD bisulfite sequencing data we recently got back from ZymoResearch.
Genome Assembly - SparseAssembler Geoduck Genomic Data, kmer=101
UPDATE 20180413
Gunzip - BGI HiSeq Geoduck Genome Sequencing Data
In preparation to run SpareAssembler, I needed to gunzip the BGI gzipped FASTQ files from 20180327.
Gunzip - Trimmed Illumina Geoduck HiSeq Genome Sequencing Data
In preparation to run SpareAssembler, I needed to gunzip the trimmed gzipped FASTQ files from 20140401.
DNA Isolation & Quantification – Geoduck larvae metagenome filter rinses
This is another attempt to isolate DNA from two more of the geoduck hatchery metagenome samples Emma delivered on 20180313.
Titrations - Yaamini’s Seawater Samples
All data is deposited in the following GitHub repo:
Titrations - Yaamini’s Seawater Samples
All data is deposited in the following GitHub repo:
TrimGalore!/FastQC/MultiQC - Illumina HiSeq Genome Sequencing Data Continued
The previous attempt at this was interrupted by a random glitch with our Mox HPC node.
Data Received - Crassostrea virginica MBD BS-seq from ZymoResearch
Received the sequencing data from ZymoResearch for the Crassostrea virginica gonad MBD DNA that was sent to them on 20180207 for bisulfite conversion, library construction, and sequencing.
FastQC/MultiQC – Illumina HiSeq Genome Sequencing Data
Since running SparseAssembler seems to be working and actually able to produce assemblies, I’ve decided I’ll try to beef up the geoduck genome assembly with the rest of our existing genomic sequencing data.
TrimGalore!/FastQC/MultiQC - Illumina HiSeq Genome Sequencing Data
Previous FastQC/MultiQC analysis of the geoduck Illumina HiSeq data (NMP.fastq.gz files) revealed a high level of overrepresented sequences, high levels of Per Base N Content, failure of Per Sequence GC Content, and a few other bad things.
FastQC/MultiQC - BGI Geoduck Genome Sequencing Data
Since running SparseAssembler seems to be working and actually able to produce assemblies, I’ve decided I’ll try to beef up the geoduck genome assembly with the rest of our existing genomic sequencing data.
Titrations - Hollie’s Seawater Samples
All data is deposited in the following GitHub repo:
Assembly - Geoduck NovaSeq using SparseAssembler kmer = 101
The prior run used a kmer size of 61, and the resulting assembly was rather poor (small N50).
Assembly - Geoduck NovaSeq using SparseAssembler (TL;DR - it worked!)
The prior attempt using SparseAssembler failed due to a kmer size that was deemed too large.
Titrations - Hollie’s Seawater Samples
All data is deposited in the following GitHub repo:
DNA Isolation & Quantification - Geoduck larvae metagenome filter rinses
Isolated DNA from two of the geoduck hatchery metagenome samples Emma delivered on 20180313 to get an idea of what type of yields we might get from these.
Titrations - Hollie’s Seawater Samples
Performed total alkalinity (TA) titrations on Hollie’s samples using our T5 Excellence titrator (Mettler Toledo) and Rondolino sample changer.
Titrations - Hollie’s Seawater Samples
Performed total alkalinity (TA) titrations on Hollie’s samples using our T5 Excellence titrator (Mettler Toledo) and Rondolino sample changer.
Samples Received - Geoduck larvae metagenome filter rinses
Received geoduck hatchery metagenome samples from Emma. These samples are intended for DNA isolation.
Assembly - Geoduck NovaSeq using SparseAssembler (failed)
Steven came across a [2012 paper in BMC Bioinformatics (“Exploiting sparseness in de novo genome assembly”)(https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-S6-S1) that utilized an assembly program we hadn’t previously encountered: SparseAssembler
Progress Report - Titrator
I’ll begin this entry with a TL;DR (becuase it’s definitely a very long read):
Ubuntu Installation - Convert Apple Xserve “bigfish” to Ubuntu
Due to hardware limitations on the Apple Xserves we have, we can’t use drives >2TB in size. “Bigfish” was set up to be RAID’d and, as such, has three existing HDDs installed.
Hardware Upgrades - USB 3.0 PCI Card and 1TB SSD in Woodpecker
Installed an additional 1TB SSD and a USB 3.0 PCI card in woodpecker to make it usable for MinION sequencing. An SSD and USB 3.0 ports are just a couple of the hardware requirements for using the sequencer. Woodpecker already satisfied other hardware requirements (processor, RAM, hard drive space).
Samples Received - Triploid Crassostrea gigas from Nisbet Oyster Company
Received a bag of Pacific oysters from Nisbet Oyster Company.
NovaSeq Assembly - The Struggle is Real - Real Annoying!
Well, I continue to struggle to makek progress on assembling the geoduck Illumina NovaSeq data. Granted, there is a ton of data (374GB!!!!), but it’s still frustrating that we can’t get an assembly anywhere…
Assembly - Geoduck Illumina NovaSeq SOAPdenovo2 on Mox (FAIL)
Trying to get the NovaSeq data assembled using SOAPdenovo2 on the Mox HPC node we have and it will not work.
Ethanol Precipitation & DNA Quantification - C. virginica MBD DNA from Yaamini
Finished the ethanol precipitation as described in the MethylMiner (Invitrogen) manual which Yaamini had previously initiated: https://yaaminiv.github.io/Virginica-MBDSeq-Day4/
NovaSeq Assembly - Trimmed Geoduck NovaSeq with Meraculous
Attempted to use Meraculous to assemble the trimmed geoduck NovaSeq data.
Titrator Setup - Functional Methods & Data Exports
I’ve been working on getting our T5 Excellence titrator (Mettler Toledo) with Rondolino sample changer (Mettler Toledo) set up and operational.
Adapter Trimming and FASTQC - Illumina Geoduck Novaseq Data
We would like to get an assembly of the geoduck NovaSeq data that Illumina provided us with.
Software Install - 10x Genomics Supernova on Mox (Hyak)
Samples Submitted - C. virginica gDNA, MBD, and MspI to Qiagen
Sent Crassostrea virginica samples to Qiagen, as part of the collaboration we have with them for testing their new bisulfite conversion kit on various reduced representation DNA.
Assembly Comparisons – Oly Assemblies Using Quast
I ran Quast to compare all of our current Olympia oyster genome assemblies.
DNA Quantification - MspI-digested Crassostrea virginica gDNA
Quantified the two MspI-digested DNA samples for the Qiagen project from earlier today with the Qubit 3.0 (ThermoFisher).
Phenol:Chloroform Extractions and EtOH Precipitations - MspI Digestions of C.virginica DNA from Earlier Today
The two MspI restriction digestions from earlier today for our project with Qiagen were subjected to phenol:chloroform cleanup and subsequent ethanol precipitations.
Restriction Digestion - MspI on Crassotrea virginica gDNA
Digested two 1.5μg aliquots of Crassostrea virginica isolated 20171211, as part of the project we’re doing with Qiagen.
DNA Quantification - C.virginica MBD-enriched DNA
Quantified Crassostrea virginica MBD-enriched DNA from earlier today for Qiagen project.
MBD Enrichment – Crassostrea virginica Sheared DNA Day 3
Continued MBD enrichment of C.virginica DNA from yesterday for Qiagen project.
MBD Enrichment – Crassostrea virginica Sheared DNA Day 2
Continued MBD enrichment for C.virginica and Qiagen project from yesterday.
MBD Enrichment - Crassostrea virginica Sheared DNA Day 1
As part of a project with Qiagen to have them try out some of our DNA with their newest DNA bisulfite conversion kit, I previously [isolated DNA from Crassotrea virginica (Eastern oyster)(2017/12/11/dna-isolation-quantification-crassotrea-virginica-mantle-gdna.html) and sheared to ~420bp.
2017
Tissue Sampling - Crassostrea virginica Tissues for Archiving
I figured it’d be prudent to collect some Eastern oyster (Crassotrea virginica) to have around the lab.
Software Install - MSMTP For Email Notices of Bash Job Completion on Emu (Ubuntu)
After I finally resolved the installation of PB Jelly on Emu (running Ubuntu 16.04), I’ve had a PB Jelly assembly running for the past two weeks! I’ve gotten tired of checking on its status (i.e. is it still running?) every day, so I dove in and figured out how to set up Emu to email me when the job is complete!
Samples Submitted - Pulverized Geoduck Tissues to Illumina for More 10x Genomics Sequencing
Continuing Illumina’s generous efforts to use our geoduck samples to test out the robustness of their emerging sequencing technologies, they have requested we send them some more geoduck tissue so that they can try to isolate higher molecular weight DNA to complete the genome sequencing efforts using the 10x genomics sequencing platform.
DNA Sonication & Bioanalzyer - C. virginica gDNA for MeDIP
I transferred 8ug (136uL) of Crassotrea virginica gDNA (isolated earlier today) to two separate 1.7mL snap cap tubes for sonication/shearing.
DNA Isolation & Quantification - Crassostrea virginica Mantle gDNA
DNA was isolated from a single adult Eastern oyster (Crassostrea virginica) for a pilot project with Qiagen to test their new DNA bisulfite conversion kit. The oyster was obtained yesterday afternoon (20171210) from the Taylo rShellfish Pioneer Square location. The oyster was stored @ 4C O/N.
Genome Assembly – Olympia Oyster Illumina & PacBio Using PB Jelly w/BGI Scaffold Assembly
After another attempt to fix PB Jelly, I ran it again.
Troubleshooting – PB Jelly Install on Emu Continued
The last “fix” didn’t fix everything.
Samples Submitted - Geoduck Tissues to Illumina for More 10x Genomics Sequencing
Continuing Illumina’s generous efforts to use our geoduck samples to test out the robustness of their emerging sequencing technologies, they have requested we send them some more geoduck tissue so that they can try to isolate higher molecular weight DNA to complete the genome sequencing efforts using the 10x genomics sequencing platform.
Troubleshooting - PB Jelly Install on Emu
I previously installed and ran PB Jelly. Despite no error messages being output, I noticed something odd during my quick post-assembly stats check: The PB Jelly numbers were identical to the input reference file. This seemed very strange and made me decide to look a bit deeper in the PB Jelly output files.
DNA Isolation & Quantification - C. virginica Gonad gDNA
I isolated DNA from the Crassotrea virginica gonad samples sent by Katie Lotterhos using the E.Z.N.A. Mollusc Kit with the following modifications:
Assembly Comparison - Oly Assemblies Using Quast
I ran Quast to compare all of our current Olympia oyster genome assemblies.
Genome Assembly - Olympia Oyster Illumina & PacBio Using PB Jelly w/BGI Scaffold Assembly
Yesterday, I ran PB Jelly using Sean’s Platanus assembly, but that didn’t produce an assembly because PB Jelly was expecting gaps in the Illumina reference assembly (i.e. scaffolds, not contigs).
Genome Assembly - Olympia Oyster Illumina & PacBio Using PB Jelly w/Platanus Assembly
Sean had previously attempted to run PB Jelly, but ran into some issues running on Hyak, so I decided to try this on Emu.
Samples Received - Tanner Crab Hemolymph in RNA Later from Pam Jensen
Pam Jensen stopped by and dropped off Tanner crab (Chionoecetes bairdi) hemolymph samples (~300) stored in RNA Later (Ambion). Samples were stored at 4C.
RNA Isolation & Quantification - Tanner crab hemolymph
We received three Tanner crab (Chionoecetes bairdi)hemolymph samples from Pam Jensen (NOAA) yesterday. From her email to Steven:
Software Installation - ALPACA on Roadrunner
List of software that needed installing to run ALPACA:
Software Crash - Olympia oyster genome assembly with Masurca on Mox
Ah, the joys of bioinformatics. I just received an email from Mox indicating that [the Masurca assembly I started 11 DAYS AGO (!!)(2017/10/19/genome-assembly-olympia-oyster-illumina-pacbio-reads-using-masurca.html) crashed.
Software Installation - PB Jelly Suite and Blasr on Emu
I followed along with what Sean previously did when installing on Emu, but it appears he didn’t install it in the shared location to make it accessible to all users. So, I’m installing it in the /home/shared/ directory.
Genome Assembly - Olympia oyster Illumina & PacBio Reads Using Redundans
Had problems with Docker and Jupyter Notebook inexplicably dying and deleting all the files in the working directory of the Jupyter Notebook (which also happened to be the volume mounted in the Docker container).
Assembly Comparison - Oly PacBio Canu: Sam vs. Sean with Quast
I recently finished an assembly of our Olympia oyster PacBio data using Canu and thought it would be interesting to compare to Sean’s Canu assembly.
FAIL - Missing Data on Owl!
Uh oh. There appears to be some data that’s been removed from Owl. I noticed this earlier when trying to look at some of Sean’s data. His data should be in a folder with his name in Owl/scaphapoda
Genome Assembly - Olympia oyster Illumina & PacBio reads using MaSuRCA
Software Installation - MaSuRCA v3.2.3 Assembler on Mox (Hyak)
Saw this tweet this morning and thought this would be good to try out for our Olympia oyster genome assemblies, as it will handle “hybrid” assemblies (i.e. short-reads and long reads):
Fail - Directory Contents Deleted!
Uh, not sure what happened here:
Genome Assembly - Olympia oyster PacBio Canu v1.6
I decided to run Canu myself, since documentation for Sean’s Canu run is a bit lacking. Additionally, it looks like he specified a genome size of 500Mbp, which is probably too small. For this assembly, I set the genome size to 1.9Gbp (based on the info in the BGI assembly report, using 17-mers for calculating genome size), which is probably on the large size.
Data Management - Convert Oly PacBio H5 to FASTQ
After working with all of this Olympia oyster genome sequencing data, I remembered that we had an old, singular PacBio SMRT cell file (from June 2013). This file didn’t seem to be included in any recent assemblies of Sean’s or mine. This is most likely because we have it in the PacBio H5 format and not in FASTQ.
Genome Assembly - Olympia oyster Redundans/Canu vs. Redundans/Racon
Decided to compare the Redundans using Canu as reference and Redundans using Racon as reference. Both reference assemblies were just our PacBio data.
Genome Assembly - Olympia Oyster Redundans with Illumina + PacBio
Redundans should assemble both Illumina and PacBio data, so let’s do that.
Genome Assembly - minimap/miniasm/racon Overview
Previously, I used the following three tools to do quick assembly of our Olympia oyster PacBio data:
Assembly Comparisons - Olympia oyster genome assemblies
— UPDATE 20171009 —
Samples Received - C.virginica gonad tissue from Katie Lotterhos
Received and stored @-80C in rack 8, row 5, column 5.
Goals - October 2017
I guess one of my primary goals is to make sure I actually write my monthly goals each month.
Genome Assembly - Olympia oyster PacBio minimap/miniasm/racon
In this GitHub Issue, Steven had suggested I try out the minimap/miniasm/racon pipeline for assembling our Olympia oyster PacBio data.
Genome Assembly - Olympia oyster PacBio minimap/miniasm/racon
In this GitHub Issue, Steven had suggested I try out the minimap/miniasm/racon pipeline for assembling our Olympia oyster PacBio data.
Genome Assembly - Olympia oyster PacBio minimap/miniasm/racon
In this GitHub Issue, Steven had suggested I try out the minimap/miniasm/racon pipeline for assembling our Olympia oyster PacBio data.
Samples Submitted - Geoduck Ctenidia to Illumina for 10x Genomics Sequencing
Continuing Illumina’s generous efforts to use our geoduck samples to test out the robustness of their emerging sequencing technologies, they have requested we send them some geoduck tissue so that they can try to complete the genome sequencing efforts using the 10x genomics sequencing platform.
Project Progress - Olympia Oyster Genome Assemblies by Sean Bennett
Data Management - Illumina Geoduck HiSeq & MiSeq Data
The HDD we received from Illumina last week only had data (i.e. fastq files) from the NovaSeq runs they performed - nothing from either the MiSeq, nor the HiSeq runs.
Goals - August 2017
Data, data, data! That is the theme for this month.
Data Received - Geoduck Genome Sequencing by Illumina
We previously sent some geoduck samples to Illumina, as part of a pilot project for them to test out a new sequencing platform. The data has finally arrived!
Sample Submission - Olympia oyster gonad RNA to Katherine Silliman @ Univ. of Chicago
Sent the following RNA to Katherine Silliman at the Univ. of Chicago for RNAseq. All RNA was isolated on 20170719, except SN-10-16, which was isolated on 20170710.
RNA Isolation - Olympia oyster gonad tissue in paraffin histology blocks
My previous go at this was a little premature - I didn’t wait for Laura to fully annotate her slides/blocks. Little did I know, the tissue was mostly visceral mass and, as such, I didn’t hit much in the way of actual gonad tissue. So, I’m redoing this, now that Grace has gone through and annotated the blocks to point out gonad tissue. SN-10-16 was sent to Katherine Silliman on 20170720.
RNA Isolation - Olympia oyster gonad tissue in paraffin histology blocks
UPDATE 20170712: The RNA I isolated below is from incorrect regions of tissue. I misunderstood exactly what this tissue was, and admittedly, jumped the gun. The tissue is actually collected from the visceral mass - which contains gonad (a small amount) and digestive gland (a large amount). The RNA isolated below will be stored in one of the Shellfish RNA boxes and I will isolate RNA from the correct regions indicated by Grace
Data Management - SRA Submission Olympia Oyster UW PacBio Data from 20170323
Submitted the FASTQ files from the UW PacBio Data from 20170323 to the [NCBI sequence read archive (SRA)(https://www.ncbi.nlm.nih.gov/sra).
Data Management – Tarball of Olympia oyster UW PacBio Data from 20170323
I’d previously attempted to archive this data set on multiple occasions, across multiple days, but network dropouts kept killing my connection to the server (Owl) and, in turn, interrupting the tarball operation.
Data Management - OLYMPIA OYSTER UW PACBIO DATA (FROM 20170323) TO NIGHTINGALES
Added UW PacBio FASTQ files to our nightingales Google Sheet for keeping track of our high-throughput sequencing projects.
Sample Annotation - Olympia oyster histology blocks (from Laura Spencer)
I’ve been asked to isolate RNA from some paraffin-embedded Olympia oyster gonad tissue.
Manuscript Re-submission - Oly Stress Response to PeerJ for Review
Data Management - Olympia oyster UW PacBio Data from 20170323
Due to other priorities, getting this PacBio data sorted and prepped for our [next gen sequencing data management plan (DMP)(https://github.com/sr320/LabDocs/wiki/Data-Management#ngs-data-management-plan) was put on the back burner. I finally got around to this, but it wasn’t all successful.
GitHub Curation
Updated a couple of GitHub Wikis:
Goals - June 2017
Well, my previous goal was to tidy up an existing manuscript and get it re-submitted to PeerJ. That’s pretty much done, as Steven will be giving a final once over and formatting the rebuttal letter prior to resubmission.
DNA Methylation Quantification - Acropora cervicornis (Staghorn coral) DNA from Javier Casariego (FIU)
Used the MethylFlash Methylated DNA Quantification Kit (Colorimetric) from Epigentek to quantify methylation in these coral DNA samples.
DNA Quantification - Acropora cervicornis (Staghorn coral) DNA from Javier Casariego (FIU)
I quantified the three samples (listed below) that I SpeedVac’d yesterday using the the Roberts Lab Qubit 3.0.
DNA Concentration - Acropora cervicornis (Staghorn coral) DNA from Javier Casariego (FIU)
Three samples (of the 62 total) that were quantified earlier today, had concentrations too low for use in the methylation assay:
DNA Quantification - Acropora cervicornis (Staghorn coral) DNA from Javier Casariego (FIU)
DNA samples received yesterday were quantified using the Roberts Lab Qubit 3.0 to improve quantification accuracy (samples provided by Javier were quantified via NanoDrop, which generally overestimates DNA concentration) prior to performing methylation assessment.
Samples Received - Olympia oyster Histology Blocks and Slides (for Laura Spencer)
Received histology blocks and slides for Laura Spencer.
Samples Received - Acropora cervicornis (Staghorn coral) DNA from Javier Casariego (FIU)
Received 62 coral (Acropora cervicornis) DNA samples from Javier Casariego at FIU.
Goals - May 2017
A day late, but definitely not a dollar short!
Manuscript Writing - Submitted!
Boom!
Manuscript - Oly GBS 14 Day Plan
For Pub-a-thon 2017, Steven has asked us to put together a 14 day plan for our manuscripts.
Data Received - Olympia oyster PacBio Data
Back in December 2016, we sent off Ostrea lurida DNA to the UW PacBio sequencing facility. This is an attempt to fill in the gaps left from the BGI genome sequencing project.
Data Management – SRA Submission Oly GBS Batch Submission
Computing - Owl Partially Restored
Heard back from Synology and they indicated I should click the “Repair” option to fix the System Partition Failed error message seen previously.
Data Management - SRA Submission Oly GBS Batch Submission Fail
[I had previously submitted the two non-demultiplexed genotype-by-sequencing (GBS) files provided by BGI to the NCBI short read archive (SRA)(2017/02/08/data-management-sra-submission-of-ostrea-lurida-gbs-fastq-files.html).
Troubleshooting - Synology NAS (Owl) Down After Update
TL;DR - Server didn’t recover after firmware update last night. “Repair” is an option listed in the web interface, but I want to confirm with Synology what will happen if/when I click that button…
Computing – Oly BGI GBS Reproducibility; fail?
OK, so things have improved since the last attempt at getting this BGI script to run and demultiplex the raw data.
Computing – Oly BGI GBS Reproducibility Fail (but, less so than last time)…
Well, my previous attempt at reproducing the demultiplexing that BGI performed was an exercise in futility. BGI got back to me with the following message:
Computing - Oly BGI GBS Reproducibility Fail
Since we’re preparing a manuscript that relies on BGI’s manipulation/handling of the genotype-by-sequencing data, I attempted to could reproduce the demultiplexing steps that BGI used in order to perform the SNP/genotyping on these samples.
FASTQC - Oly BGI GBS Raw Illumina Data Demultiplexed
Last week, I ran the two raw FASTQ files through FastQC. As expected, FastQC detected “errors”. These errors are due to the presence of adapter sequences, barcodes, and the use of a restriction enzyme (ApeKI) in library preparation. In summary, it’s not surprising that FastQC was not please with the data because it’s expecting a “standard” library prep that’s already been trimmed and demultiplexed.
FASTQC - Oly BGI GBS Raw Illumina Data
In getting things prepared for the manuscript we’re writing about the Olympia oyster genotype-by-sequencing data from BGI, I felt we needed to provide a FastQC analysis of the raw data (since these two files are what we submitted to the NCBI short read archive) to provide support for the Technical Validation section of the manuscript.
Goals - March 2017
Goal, singular: Get Oly GBS manuscript completed/submitted.
Manuscript Writing - More “Nuances” Using Authorea
I previously highlighted some of the issues I was having using Authorea.com as an writing platform.
Data Received - Jay’s Coral RADseq and Hollie’s Geoduck Epi-RADseq
Jay received notice from UC Berkeley that the sequencing data from his coral RADseq was ready. In addition, the sequencing contains some epiRADseq data from samples provided by Hollie Putnam. See his notebook for multiple links that describe library preparation (indexing and barcodes), sample pooling, and species breakdown.
Data Management - SRA Submission of Ostrea lurida GBS FASTQ Files
Prepared a short read archive (SRA) submission for archiving our Olympia oyster genotype-by-sequencing (GBS) data in NCBI. This is in preparation for submission of the mansucript we’re putting together.
Goals - February 2017
First goal is to be the first person in lab to post their goals each month. Props to one of our new grad students, Yaamini Venkataraman on beating me this month!
Manuscript Writing - The “Nuances” of Using Authorea
I’m currently trying to write a manuscript covering our genotype-by-sequencing data for the Olympia oyster using the Authorea.com platform and am encountering some issues that are a bit frustrating. Here’s what’s happening (and the ways I’ve managed to get around the problems).
Sample Submission – Geoduck gDNA for Illumina Pilot Sequencing Project
Sent 10μg of the geoduck gDNA I isolated earlier today to Illumina on dry ice via FedEx Standard Overnight service.
DNA Isolation - Geoduck gDNA for Illumina-initiated Sequencing Project
We were previously approached by Cindy Lawley (Illumina Market Development) for possible participation in an Illumina product development project, in which they wanted to have some geoduck tissue and DNA on-hand in case Illumina green-lighted the use of geoduck for testing out the new sequencing platform on non-model organisms. Well, guess what, Illumina has give the green light for sequencing our geoduck! However, they need at least 4μg of gDNA, so I’m isolating more.
Data Management - Replacement of Corrupt BGI Oly Genome FASTQ Files
Previously, Sean and Steven identified two potentially corrupt FASTQ files. I contacted BGI about getting replacement files and they informed me that all versions of the FASTQ files they have delivered on three separate occasions are all the same file (despite having different file names). As such, I could use one of these versions to replace the corrupt FASTQ files. So, that’s what I did!
Goals - January 2017
One of the long-running goals I’ve had is to get this Oly GBS data taken care of and out the door to publication. I think I will finally succeed with this, with the help of Pub-A-Thon. Don’t get too excited, it’s not what you think. It is not the drinking extravaganza that the name implies. Instead, it’s a “friendly” lab competition to get some scientific publications assembled and submitted.
2016
Data Management - Geoduck RRBS Data Integrity Verification
Yesterday, I downloaded the Illumina FASTQ files provided by Genewiz for Hollie Putnam’s reduced representation bisulfite geoduck libraries. However, Genewiz had not provided a checksum file at the time.
Data Received - Geoduck RRBS Sequencing Data
Hollie Putnam prepared some reduced representation bisulfite Illumina libraries and had them sequenced by Genewiz.
Sample Submission - Geoduck Tissue & gDNA for Illumina Pilot Sequencing Project
Sent the following samples to Illumina for possible selection in a new pilot sequencing platform they’re working on.
DNA Isolation - Geoduck gDNA for Potential Illumina-initiated Sequencing Project
We were approached by Cindy Lawley (Illumina Market Development) yesterday to see if we’d be able to participate in some product development. We agreed and need some geoduck DNA to send them, in case she’s able to get our species greenlighted for use.
Sample Submission - Geoduck Reduced Representation Bisulfite Pooled Libraries
Hollie Putnam asked me to help her get samples ready for submission for Illumina sequencing at Genewiz.
Sample Submission - Ostrea lurida gDNA for PacBio Sequencing
Submitted 10μg (30.7μL) of the O.lurida gDNA I isolated on 20161214 to the UW PacBio facility - Order #450.
Data Management - Integrity Check of Final BGI Olympia Oyster & Geoduck Data
After completing the downloads of these files from BGI, I needed to verify that the downloaded copies matched the originals. Below is a Jupyter Notebook detailing how I verified file integrity via MD5 checksums. It also highlights the importance of doing this check when working with large sequencing files (or, just large files in general), as a few of them had mis-matching MD5 checksums!
DNA Isolation - Ostrea lurida DNA for PacBio Sequencing
In an attempt to improve upon the partial genome assembly we received from BGI, we will be sending DNA to the UW PacBio core facility for additional sequencing.
Data Managment - Trim Output Cells from Jupyter Notebook
Last week I downloaded the final BGI data files and assemblies for Olympia oyster and geoduck genome sequencing projects. However, the output from the download command made the Jupyter Notebook files too large to view and/or upload to GitHub. So, I had to trim the output cells from that notebook in order to render it usable/viewable.
Data Management - Download Final BGI Genome & Assembly Files
We received info to download the final data and genome assembly files for geoduck and Olympia oyster from BGI.
Goals - December 2016
Well, I’ve finally progressed with the Olly GBS analysis!
Data Analysis - Continued O.lurida Fst Analysis from GBS Data
Continued the analysis I started the other day. Still following Katherine Silliman’s notebook for guidance.
Computing - An Excercise in Futility
Trying to continue my Oly GBS analsyis from the other day and follow along with Katherine Silliman’s notebook
Data Analysis - Initial O.lurida Fst Determination from GBS Data
Finally running some analysis on the output from my PyRad analysison 20160727.
Data Management - Tracking O.lurida FASTQ File Corruption
UPDATE 20170104 - These two corrupt files have been replaced with non-corrupt files.
Computer Management - Additional Configurations for Reformatted Xserves
Sean got the remaining Xserves configured to run independently from the master node of the cluster they belonged to and installed OS X 10.11 (El Capitan).
Data Management - Modify Eagle/Owl Cloud Sync Account
Re-examining our backup options for our two Synology servers (Eagle & Owl), I realized that they were both backing up to the just my account on UW’s unlimited Google Drive storage.
Computing - Retrieve data from Amazon EC2 Instance
I had an existing instance that still had data on it from my PyRad analysis on 20160727 that I needed to retrieve.
Goals - November 2016
Well, I’m serious this time. My goal for this month is to complete the Oly GBS data analysis and, get the data sets and data analysis prepared/placed in satisfactory repositories in preparation for publication in Scientific Data.
Data Management – Geoduck Small Insert Library Genome Assembly from BGI
Received another set of Panopea generosa genome assembly data from BGI back in May! I neglected to create MD5 checksums, as well as a readme file for this data set! Of course, I needed some of the info that the readme file should’ve had and it wasn’t there. So, here’s the skinny…
Goals - October 2016
Last month’s goals, as it turns out, were way too ambitious. This month’s goal will be to get the Oly GBS data analysis fully completed (currently have individuals data, but need summary of the three populations data). I’ll also get the data sets and data analysis prepared/placed in satisfactory repositories in preparation for publication in Scientific Data.
Data Received – Jay’s Coral epiRADseq - Not Demultiplexed
Previously downloaded Jay’s epiRADseq data that was provided by the Genomic Sequencing Laboratory at UC-Berkeley. It was provided already demultiplexed (which is very nice of them!). To be completionists on our end, we requested the non-demultiplexed data set.
Oyster Sampling - Olympia Oyster OA Populations at Manchester
I helped Katherine Silliman with her oyster sampling today from her ocean acidification experiment with Olympia oysters (Ostrea lurida) at the Kenneth K. Chew Center for Shellfish Research & Restoration, which is housed at the NOAA Northwest Fisheries Science Center at Manchester in a partnership with the [Puget Sound Restoration Fund (PSRF)(http://www.restorationfund.org/). We sampled the following tissues and stored in 1mL RNAlater:
Data Received - Jay’s Coral epiRADseq
We received notice that Jay’s coral (Porites spp) epiRADseq data was available from the Genomic Sequencing Laboratory at UC-Berkeley.
Goals - September 2016
Whoops! It’s already September 6th! The 1st of the month came and went without me noticing.
Data Management - Synology Cloud Sync to UW Google Drive
After a bit of a scare this weekend (Synology DX513 expansion unit no longer detected two drives after a system update - resolved by powering down the system and rebooting it), we revisited our approach for backing up data.
Server HDD Failure – Owl
Noticed that Owl (Synology DS1812+ server) was beeping.
Manuscript Submission - Oly Stress Response to PeerJ for Review
Submitted the following manuscript to PeerJ for peer review:
Data Analysis - fastStructure Population Analysis of Oly GBS PyRAD Output
After some basal readings about what Fst is (see notebook below for a definition and reference), I decided to try to use fastStructure to analyze the PyRAD output from 20160727.
Computing - Amazon EC2 Cost “Analysis”
I recently moved some computing jobs over to Amazon’s Elastic Cloud Computing (EC2) in attempt to avoid some odd computing issues/errors I kept encountering on our lab computers (Apple Xserve 3,1).
Goals - August 2016
- Complete Olympia oyster GBS data analysis - Progress has actually been made! After many struggles, I managed to get a PyRad analysis of the entire data set to complete. Now, I just have to figure out what to do with the output files…
Data Analysis - PyRad Analysis of Olympia Oyster GBS Data
Computing - Not Enough Power!
Well, I tackled the storage space issue by expanding the EC2 Instance to have a 1000GB of storage space. Now that that’s no longer a concern, it turns out I’m running up against processing/memory limits!
Computing - Amazon EC2 Instance Out of Space?
Running PyRad analysis on the Olympia oyster GBS data. PyRad exited with warnings about running out of space. However, looking at free disk space on the EC2 Instance suggests that there’s still space left on the disk. Possibly PyRad monitors the expected disk space usage during analysis to verify there will be sufficient disk space to write to? Regardless, will expand EC2 volume instance to a larger size…
Computing - A Very Quick “Guide” to Amazon EC2 Continued
Yesterday’s post ended with me trying to mount a S3 bucket to my EC2 instance using s3fs-fuse.
Dissection - Frozen Geoduck & Pacific Oyster
We’re working on a project with Washington Department of Natural Resources’ (DNR) Micah Horwith to identify potential proteomic biomarkers in geoduck (Panopea generosa) and Pacific oyster (Crassostrea gigas). One aspect of the project is how to best conduct sampling of juvenile geoduck (Panopea generosa) and Pacific oyster (Crassostrea gigas) to minimize changes in the proteome of ctenidia tissue during sampling. Generally, live animals are shucked, tissue dissected, and then the tissue is “snap” frozen. However, Micah’s crew will be collecting animals from wild sites around Puget Sound and, because of the remote locations and the means of collection, will have limited tools and time to perform this type of sampling. Time is a significant component that will have great impact on proteomic status in each individual.
Filter Replacement - Xserve Server Rack Enclosure
Replaced the filters on the rack enclosure that houses the Apple Xserver server blades we have in the lab.
Computing - The Very Quick “Guide” to Amazon Web Services Cloud Computing Instances (EC2)
This all takes a surprisingly long time to set up.
DNA Methylation Quantification - Coral DNA from Jose M. Eirin-Lopez (Florida International University)
Ran the coral DNA I quantified on 20160630 through the MethylFlash Methylated DNA Quantification Kit [Colorimetric] (Epigentek) kit to quantify global methylation.
Goals - July 2016
Unfortunately, most of this month’s goals are the same as last months!
DNA Quantification - Coral DNA from Jose M. Eirin-Lopez (Florida International University)
Quantified the DNA we received from Jose on 20160615 using the Qubit 3.0 Flouorometer (ThermoFisher) with the dsDNA Broad Range (BR) Kit according to the manufacturer’s protocol. Used 1μL of each sample.
Samples Received - Coral DNA from Jose M. Eirin-Lopez (Florida International University)
Steven received these coral DNA samples today. Here’s his post on Google Plus (stored @ 4C in FTR 213):
Docker - VirtualBox Defaults on OS X
I noticed a discrepancy between what system info is detected natively on Roadrunner (Apple Xserve) and what was being shown when I started a Docker container.
Docker - One liner to create Docker container
One liner to create Docker container for Jupyter notebook usage and data analysis on roadrunner (Xserve):
RAM Upgrade - Roadrunner (Apple Xserve) to 48GB RAM
We received the new 48GB RAM set we ordered from Other World Computing for the Apple Xserve (roadrunner) that I installed El Capitan (OS X 10.11.5) on two weeks ago.
Goals - June 2016
Current goals are as follows:
Docker - Improving Roberts Lab Reproducibility
In an attempt at furthering our lab’s abilities to maximize our reproducibility, I’ve been working on developing an all-encompassing Docker image. Docker is a type of virtual machine (i.e. a self-contained computer that runs within your computer). For the Roberts Lab, the advantage of using Docker is that the Docker images can be customized to run a specific suite of software and these images can then be used by any other person in the lab (assuming they can run Docker on their particular operating system), regardless of operating system. In turn, if everyone is using the same Docker image (i.e. the same virtual machine with all the same software), then we should be able to reproduce data analyses more reliably, due to the fact that there won’t be differences between software versions that people are using. Additionally, using Docker greatly simplifies the setup of new computers with the requisite software.
Computer Setup - Cluster Node003 Conversion
Here’s an overview of some of the struggles getting node003 converted/upgraded to function as an independent computer (as opposed to a slave node in the Apple computer cluster).
Data Analysis - Oly GBS Data Using Stacks 1.37
This analysis ran (or, more properly, was attempted) for a couple of weeks and failed a few times. The failures seemed to be linked to the external hard drive I was reading/writing data to. It continually locked up, leading to “Segmentation fault” errors.
Data Management - Olympia Oyster Small Insert Library Genome Assembly from BGI
Received another set of Ostrea lurida genome assembly data from BGI. In this case, it’s data assembled from the small insert libraries they created for this project.
GBS Frustrations
This isn’t really a notebook entry - it’s more of a traditional blog post.
Goals - May 2016
Well, I guess the first goal is to remember to be more consistent about writing monthly goals…
SRA Release - Transcriptomic Profiles of Adult Female & Male Gonads in Panopea generosa (Pacific geoduck)
The RNAseq data that [I previously submitted to NCBI short read archive (SRA)(2016/03/24/sra-submission-transcriptomic-profiles-of-adult-female-male-gonads-in-panopea-generosa-pacific-geoduck.html) has been released to the public today. Here are the various links for the project:
Data Management - O.lurida Raw BGI GBS FASTQ Data
BGI had previously supplied us with demultiplexed GBS FASTQ files. However, they had not provided us with the information/data on how those files were created. I contacted them and they’ve given us the two original FASTQ files, as well as the library index file and corresponding script they used for demultiplexing all of the files. The Jupyter (iPython) notebook below updates our checksum and readme files in our server directory that’s hosting the files: http://owl.fish.washington.edu/nightingales/O_lurida/20160223_gbs/
Computing - Speed Benchmark Comparisons Between Local, External, & Server Files
I decided to run a quick test to see what difference in speed (i.e. time) we might see between handling files that are stored locally, on an external hard drive (HDD), or on our server (Owl).
Data Analysis - Subset Olympia Oyster GBS Data from BGI as Single Population Using PyRAD
Attempting to get some sort of analysis of the Ostrea lurida GBS data from BGI, particularly since the last run at it using Stacks crashed (literally) and burned (not literally).
Data Management - Concatenate FASTQ files from Oly MBDseq Project
Steven requested I concatenate the MBDseq files we received for this project:
Data Analysis - Oly GBS Data from BGI Using Stacks
UPDATE (20160418) : I’m posting this more for posterity, as Stacks continually locked up at both the “ustacks” and “cstacks” stages. These processes would take days to run (on the full 96 samples) and then the processes would become “stuck” (viewed via the top command in OS X).
Software Install - samtools-0.1.19 and stacks-1.37
Getting ready to analyze our Ostrea lurida genotype-by-sequencing data and wanted to use the Stacks software.
SRA Submission – Genome sequencing of the Olympia oyster (Ostrea lurida)
Adding our Olympia oyster genome sequencing (sequencing done by BGI) to the NCBI Sequence Read Archive (SRS). The current status can be seen in the screen cap below. Release date is set for a year from now, but will likely bump it up. Need Steven to review the details of the submission (BioProject, Experiment descriptions, etc.) before I initiate the public release. Will update this post with the SRA number once we receive it.
SRA Submission – Genome sequencing of the Pacific geoduck (Panopea generosa)
Adding our geoduck genome sequencing (sequencing done by BGI) to the NCBI Sequence Read Archive (SRS). The current status can be seen in the screen cap below. Release date is set for a year from now, but will likely bump it up. Need Steven to review the details of the submission (BioProject, Experiment descriptions, etc.) before I initiate the public release. Will update this post with the SRA number once we receive it.
SRA Submission - Transcriptomic Profiles of Adult Female & Male Gonads in Panopea generosa (Pacific geoduck).
RNAseq experiment, which is part of a larger project that involves characterizing geoduck gonad development across multiple stages: histologically, proteomically, and transcriptomically. Initial sample collection performed by Grace Crandall.
SRA Submission - Individual Transcriptomic Profiles of C.gigas Before & After Heat Shock
RNA-seq experiment conducted by Claire in 2013.
Data Management - SRA Submission Overview
We have an enormous backlog of high-throughput sequencing files (641 FASTQ files, to be exact) that we need/want to get added to the NCBI Sequence Read Archive (SRA).
Data Management - O. lurida genotype-by-sequencing (GBS) data from BGI
We received a hard drive from BGI on 20160223 (while I was out on paternity leave) containing the Ostrea lurida GBS data.
Data Received - Initial Geoduck Genome Assembly from BGI
The initial assembly of the Ostrea lurida genome is available from BGI. Currently, we’ve stashed it here:
Data Received - Initial Olympia oyster Genome Assembly from BGI
The initial assembly of the Ostrea lurida genome is available from BGI. Currently, we’ve stashed it here:
Data Management - O.lurida 2bRAD Dec2015 Undetermined FASTQ files
An astute observation by Katherine Silliman revealed that the FASTQ files I had moved to our high-throughput sequencing archive on our server Owl, only had two FASTQ files labeled as “undetermined”. Based on the number of lanes we had sequenced, we should have had many more. Turns out that the undetermined FASTQ files that were present in different sub-folders of the Genewiz project data were not uniquely named. Thus, when I moved them over (via a bash script), the undetermined files were continually overwritten, until we were left with only two FASTQ files labeled as undetermined.
Data Management - High-throughput Sequencing Data
We’ve had a recent influx of sequencing data, which is great, but it created a bit of a backlog documenting what we’ve received.
Data Received - Ostrea lurida MBD-enriched BS-seq
Received the Olympia oyster, MBD-enriched BS-seq sequencing files (50bp, single read) from ZymoResearch (submitted 20151208). Here’s the sample list:
Data Received - Ostrea lurida genome sequencing files from BGI
Downloaded data from the BGI project portal to our server, Owl, using the Synology Download Station. Although the BGI portal is aesthetically nice, it’s set up poorly for bulk downloads and took a few tries to download all of the files.
Data Received - Panopea generosa genome sequencing files from BGI
Downloaded data from the BGI project portal to our server, Owl, using the Synology Download Station. Although the BGI portal is aesthetically nice, it’s set up poorly for bulk downloads and took a few tries to download all of the files.
Data Analysis - Identification of duplicate files on Eagle
Recently, we’ve been bumping into our storage limit on Eagle (our Synology DS413):
Data Received - Bisulfite-treated Illumina Sequencing from Genewiz
2015
Data Received - Oly 2bRAD Illumina Sequencing from Genewiz
The data was made available to use on 20151224 and took two days to download.
Sample Submission - BS-seq Library Pool to Genewiz
Pooled 10ng of each of the libraries prepared yesterday with [TruSeq DNA Methylation Library Kit (Illumina)(https://github.com/sr320/LabDocs/blob/master/protocols/Commercial_Protocols/Illumina_truseq-dna-methylation-library-prep-guide-15066014-a.pdf) for sequencing at Genewiz.
Illumina Methylation Library Quantification - BS-seq Oly/C.gigas Libraries
Re-quantified the libraries that were completed yesterday using the Qubit3.0 dsDNA HS (high sensitivity) assay because the library concentrations were too low for the normal broad range kit.
Illumina Methylation Library Construction - Oly/C.gigas Bisulfite-treated DNA
Took the bisulfite-treated DNA from 20151218 and made Illumina libraries using the [TruSeq DNA Methylation Library Kit (Illumina)(https://github.com/sr320/LabDocs/blob/master/protocols/Commercial_Protocols/Illumina_truseq-dna-methylation-library-prep-guide-15066014-a.pdf).
Bioanalyzer - Bisulfite-treated Oly/C.gigas DNA
Following the guidelines of the [TruSeq DNA Methylation Library Prep Guide (Illumina)(https://github.com/sr320/LabDocs/blob/master/protocols/Commercial_Protocols/Illumina_truseq-dna-methylation-library-prep-guide-15066014-a.pdf), I ran 1μL of each sample on an RNA Pico 6000 chip on the Seeb Lab’s Bioanalyzer 2100 (Agilent) to confirm that bisulfite conversion from earlier today worked.
Reagent Prep - RNA Pico 6000 Ladder
Prepared the RNA Pico 6000 ladder (Agilent) according to the manufacturer’s protocol:
Bisulfite Treatment - Oly Reciprocal Transplant DNA & C.gigas Lotterhos DNA for BS-seq
After confirming that the DNA available for this project looked good, I performed bisulfite treatment on the following gDNA samples:
Agarose Gel - Oly gDNA for BS-seq Libraries, Take Two
The gel I ran earlier today looked real rough, due to the fact that I didn’t bother to equalize loading quantities of samples (I just loaded 1μL of all samples regardless of concentration). So, I’m repeating it using 100ng of DNA from all samples.
Agarose Gel - Oly gDNA for BS-seq Libraries
Ran 1μL of each sample from yesterday’s DNA isolation on a 0.8% agarose, low-TAE gel, stained with ethidium bromide.
DNA Isolation - Oly gDNA for BS-seq
Need DNA to prep our own libraries for bisulfite-treated high-throughput sequencing (BS-seq).
Sample Submission - 2bRAD Libraries for Genewiz
Pooled the libraries into a single sample for sequencing on Illumina HiSeq2500 by Genewiz.
Samples Received - C.gigas Tissue & DNA from Katie Lotterhos
Received 6 samples from Katie today. The box was labeled and stored @ -20C.
Sample Submission - Olympia oyster MBD-enriched DNA to ZymoResearch
We opted to go with ZymoResearch for this project because they could do the bisulfite treatment and finish the sequencing by the end of January.
Sample Submission - Additional Olympia Oyster gDNA for Genome Sequencing @ BGI
Yep, BGI still needs more gDNA for the Olympia oyster genome sequencing project. Samples have been quantified via dye-based fluorescence, as opposed to the NanoDrop, so our quantities should be more accurate and in-line with what BGI will also find.
Sample Submission - Additional Geoduck gDNA for Genome Sequencing @ BGI
Yep, BGI still needs more gDNA for the geoduck genome sequencing project. Samples have been quantified via dye-based fluorescence, as opposed to the NanoDrop, so our quantities should be more accurate and in-line with what BGI will also find.
Data Storage - Synology DX513
Running a bit low on storage on Owl (Synology DS1812+) and we will be receiving a ton of data in the next few months, so we purchased a Synology DX513. It’s an expansion unit designed specifically for seamlessly expanding our existing storage volume on Owl.
Sample Submission - Oly Oyster Bay Tissues for GBS
Sent Olympia oyster tissue samples to BGI for genotype-by-sequencing (GBS). Tissues were shipped FedEx standard overnight on dry ice.
Samples Received - Oly Tissue & DNA from Katherine Silliman
Samples were stored @ -20C in FTR 209.
DNA Quality Assessment - Geoduck & Olympia Oyster gDNA
Have three separate sets of geoduck & olympia oyster gDNA that need to be run on gels before sending to BGI for genome sequencing:
DNA Isolation - Olympia Oyster Outer Mantle gDNA
Isolated additional gDNA for the genome sequencing. To try to improve the quality (260/280 & 260/230 ratios) of the gDNA, I added a chloroform step after the initial tissue homogenization.
DNA Isolation - Geoduck Ctenidia gDNA
Isolated additional gDNA for the genome sequencing. In an attempt to obtain better yields, I used ctenidia (instead of adductor muscle). Additionally, to try to improve the quality (260/280 & 260/230 ratios) of the gDNA, I added a chloroform step after the initial tissue homogenization.
Phenol-Chloroform DNA Cleanup - Geoduck gDNA
The gDNA I extracted on 20151104 didn’t look great on the NanoDrop so I decided to perform a phenol-chloroform cleanup to see if I could improve the NanoDrop1000 absorbance spectrum and, in turn, the quality of the gDNA.
Phenol-Chloroform DNA Cleanup - Olympia Oyster gDNA
The gDNA I extracted on 20151104 didn’t look great on the NanoDrop so I decided to perform a phenol-chloroform cleanup to see if I could improve the NanoDrop1000 absorbance spectrum and, in turn, the quality of the gDNA.
DNA Isolation - Geoduck Adductor Muscle gDNA
Since we still don’t have sufficient gDNA for the full scope of the genome sequencing, I isolated more gDNA.
DNA Isolation - Olympia Oyster Outer Mantle gDNA
Since we still don’t have sufficient gDNA for the full scope of the Olympia oyster genome sequencing, I isolated more gDNA.
DNA Quantification - MBD-enriched Olympia oyster DNA
Quantified the MBD enriched samples prepped over the last two days: MBD enrichment, EtOH precipiation.
Ethanol Precipitation - Olympia oyster MBD
Precipitated the MBD enriched DNA from yesterday according to the MethylMiner Methylated DNA Enrichment Kit (Invitrogen) protocol.
MBD Enrichment - Sonicated Olympia Oyster gDNA
Olympia oyster gDNA that had previously been sonicated and fragmented was enriched for the methylated fragments using the MethylMiner Methylated DNA Enrichment Kit (Invitrogen).
DNA Sonication - Oly gDNA for MBD
In preparation for MBD enrichment, fragmented Olympia oyster gDNA with a target size of ~350bp.
qPCR – Oly RAD-Seq Library Quantification
After yesterday’s attempt at quantification revealed insufficient dilution of the libraries, I repeated the qPCRs using 1:100000 dilutions of each of the libraries. Used the KAPA Illumina Quantification Kit (KAPA Biosystems) according to the manufacturer’s protocol.
qPCR - Oly RAD-Seq Library Quantification
The final step before sequencing these 2bRAD libraries is to quantify them. Used the KAPA Illumina Quantification Kit (KAPA Biosystems) according to the manufacturer’s protocol.
Gel Extraction - Oly RAD-Seq Prep Scale PCR
Extracted the PCR products from the gel slices from 20151113 using the QIAQuick Gel Extraction Kit (Qiagen) according to the manufacturer’s protocol. Substituted MiniElute columns so that I could elute with a smaller volume than what is used in the QIAQuick standard protocol.
DNA Quality Assessment - Geoduck, Oly & Oly 2SN
I recently ran gDNA isolated for geoduck and Olympia oyster genome sequencing, as well as gDNA isolated from the Olympia oyster reciprocal transplant experiment out on a Bioanalyzer (Agilent) using the DNA 12000 chips. The results from the chip were a bit confusing and difficult to assess exactly what was going on with the DNA.
PCR – Oly RAD-seq Prep Scale PCR
Continuing with the RAD-seq library prep. Following the Meyer Lab 2bRAD protocol. After determining the minimum number of PCR cycles to run to generate a visible, 166bp band on a gel yesterday, ran a full library “prep scale” PCR.
PCR – Oly RAD-seq Test-scale PCR
Continuing with the RAD-seq library prep. Following the Meyer Lab 2bRAD protocol.
DNA Quantification & Quality Assessment - Oly 2SN gDNA
Comparison of three different approaches to using the E.Z.N.A. Mollusc Kit:
DNA Quantification & Quality Assessment - Geoduck & Oly gDNA
Quantified the following samples with the Roberts Lab NanoDrop1000 (ThermoFisher) and assessed DNA integrity on the Seeb Lab Bioanalyzer 2100 (Agilent) using the DNA 12000 chip assay:
DNA Isolations – Oly Fidalgo 2SN Ctenidia
Isolated DNA from 24 2SN ctenidia samples from Friday’s sampling (#32 - 55). Samples were thawed at RT.
Oyster Sampling - Oly Fidalgo 2SN, 2HL, 2NF Reciprocal Transplants Final Samplings
The remaining Olympia oysters from Jake Heare’s reciprocal transplant experiment have been retrieved from field sites and are awaiting sampling. The oysters have been stored in the cold room (temp?) for 15 days so far.
DNA Isolation – Geoduck & Olympia Oyster
Amazingly, we need more gDNA for the two genome sequencing projects (geoduck and Olympia oyster). Used geoduck adductor muscle sample from Box 1 of the geoduck samples collected by Brent & Steven on 20150811. Used Olympia oyster ctenidia from Box 1 of adductor muscle sample collected by Brent & Steven on 20150812.
Adaptor Ligation – Oly AlfI-Digested gDNA for RAD-seq
Continued to follow the 2bRAD protocol (PDF) developed by Eli Meyer’s lab.
Restriction Digest – Oly gDNA for RAD-seq w/AlfI
The previous attempt at making these RAD libraries failed during the prep-scale PCR, likely due to a discrepancy in the version of the Meyer Lab protocol I was following, so I have to start at the beginning to try to make these libraries again.
Troubleshooting - Oly RAD-seq
After the failure of the prep-scale PCR for the RAD library construction, Katherin Silliman pointed out [a potential problem (too much dNTPs)(https://onsnetwork.org/kubu4/2015/10/13/pcr-oly-rad-seq-prep-scale-pcr/#comments). This was odd because I was following the Meyer Protocol and I used what was indicated.
DNA Isolations - Fidalgo 2SN Reciprocal Transplants Final Samplings
The remaining Olympia oysters from Jake Heare’s reciprocal transplant experiment have been retrieved from field sites and are awaiting sampling. The oysters have been stored in the cold room (temp?) for 6 days so far.
PCR - Oly RAD-seq Prep Scale PCR
Continuing with the RAD-seq library prep. Following the Meyer Lab 2bRAD protocol.
PCR - Oly RAD-seq Test-scale PCR
Continuing with the RAD-seq library prep. Following the Meyer Lab 2bRAD protocol.
Adaptor Ligation – Oly AlfI-Digested gDNA for RAD-seq
Continued to follow the 2bRAD protocol (PDF) developed by Eli Meyer’s lab.
Restriction Digest – Oly gDNA for RAD-seq w/AlfI
Previously initiated the RAD-seq procedure for the sample set described below. However, the test scale PCR yielded poor results. Katherine Silliman suggested that the poor performance of the test scale PCR was likely due to low numbers of adaptor-ligated fragments. Since the input DNA is so degraded, I’ve repeated this using 9μg of input DNA (instead of the recommended 1.2μg). This should increase the number of available cleavage sites for AlfI, thus improving the number of available ligation sites for the adaptors.
Sample Submission - Additional Geoduck gDNA for Genome Sequencing @ BGI
Previous shipment of gDNA proved to be of insufficient quantity when assessed by BGI, so needed to isolate more.
Sample Submission - Additional Olympia Oyster gDNA for Genome Sequencing @ BGI
Previous shipment of gDNA proved to be of insufficient quantity when assessed by BGI, so needed to isolate more.
Agarose Gel - Geoduck & Olympia Oyster gDNA
Needed to assess the integrity of the newest gDNA isolated for the two genome sequencing projects: Geoduck gDNA from earlier today and Olympia oyster gDNA from 20151002.
DNA Quantification - Pooled geoduck gDNA
Pooled the gDNA samples from earlier today & from 20151002.
gDNA Isolation - Geoduck Adductor Muscle
My isolation on Friday didn’t yield a sufficient quantity of gDNA for the additional DNA needed for the geoduck genome sequencing project. Used two adductor muscles (Box 1) samples collected by Brent & Steven on 20150811.
DNA Isolation - Geoduck & Olympia Oyster
Amazingly, we need more gDNA for the two genome sequencing projects (geoduck and Olympia oyster). Used geoduck “foot 1” sample from Box 1 of the foot samples collected by Brent & Steven on 20150811. Used Olympia oyster adductor muscle from Box 1 of adductor muscle sample collected by Brent & Steven on 20150812.
PCR - Oly RAD-seq Test-scale PCR
Yesterday’s test scale PCR failed to produce any bands in any samples (expected size of ~166bp). This is not particularly surprising, due to the level of degradation in these samples. As such, repeated the test scale PCR, but increased the number of cycles.
PCR - Oly RAD-seq Test-scale PCR
Continuing with the RAD-seq library prep. Following the Meyer Lab 2bRAD protocol.
Adaptor Ligation - Oly AlfI-Digested gDNA for RAD-seq
Yesterday’s AlfI over night restriction digest was heat inactivated by heating @ 65C for 10mins. Samples were stored on ice.
Goals - October 2015
I’d review last month’s goals, but I completely forgot to post them!
Restriction Digest - Oly gDNA for RAD-seq w/AlfI
Used a subset (10 samples) from the Ostrea lurida gDNA isolated 20150916 to prepare RAD libraries. This will be done to assess whether or not these samples, which appear to be heavily degraded, are viable for RAD-seq.
Sample Submission - Olympia Oyster gDNA for Genome Sequencing @ BGI
Shipped the pooled gDNA we’ve been accumulating to BGI to initiate the Olympia oyster genome sequencing project.
Sample Submission - Geoduck gDNA for Genome Sequencing @ BGI
Shipped the pooled gDNA we’ve been accumulating to BGI to initiate the geoduck genome sequencing project.
Uninterruptible Power Supplies (UPS)
A new UPS we installed this week for our qPCR machine (Opticon2 - BioRad) to handle power surges and power outages doesn’t seem to be working properly. With the qPCR machine (and computer and NanoDrop1000) plugged into the “battery” outlets on the UPS, this is what happens when the Opticon goes through a heating cycle:
Agarose Gel - Geoduck & Olympia oyster gDNA Integrity Check
Ran a 0.8% agarose, 1x modified TAE gel (w/EtBr) with geoduck and Olympia oyster gDNA that was precipitated earlier today. Used 5μL of each sample (~500ng).
Ethanol Precipitation - Geoduck & Olympia oyster gDNA
Pooled all of the geoduck gDNA from all the previous geoduck isolations for the Geoduck Genome Sequencing Project and pooled all of the Ostrea lurida gDNA from previous Ostrea lurida isolations for the Olympia Oyster Genome Sequencing Project.
Agarose Gel - Olympia oyster Whole Body gDNA Integrity Check
Ran the gDNA isolated yesterday from Ostrea lurida whole body on a 0.8% modified TAE gel (w/EtBr) to assess gDNA integrity. Used 1μL of each sample.
DNA Isolation - Olympia oyster whole body
Continued the extractions that Steven began yesterday and this morning using the E.Z.N.A. Mollusc DNA Kit (Omega Bio-Tek) after the RNase treatment @ 70C.
Agarose Gel – Geoduck gDNA Integrity Check
Ran 0.8% agarose 1x modified TAE gel stained with EtBr to assess the integrity of geoduck gDNA isolated earlier today.
Genomic DNA Isolation – Geoduck Adductor Muscle & Foot
Just need a tad bit more gDNA for the geoduck genome sequencing project with BGI. Currently have ~69 and need a minimum of 73μg.
Agarose Gel - Geoduck & Olympia Oyster gDNA Integrity Check
Ran 0.8% agarose 1x modified TAE gel stained with EtBr to assess the integrity of geoduck gDNA and Olympia oyster gDNA isolated earlier today.
Genomic DNA Isolation – Olympia oyster adductor musle & mantle
Isolated gDNA from Ostrea lurida (Olympia oyster) adductor muscle & mantle samples collected by Brent & Steven on 20150812 using DNAzol (Molecular Research Center) according to the manufacturer’s protocol, with the following adjustments:
Genomic DNA Isolation – Geoduck Adductor Muscle & Foot
Isolated gDNA from Panopea generosa (geoduck) adductor muscle & foot samples collected by Brent & Steven on 20150811 using the DNAzol (Molecular Research Center) according to the manufacturer’s protocol, with the following adjustments:
Agarose Gel - Geoduck & Olympia Oyster gDNA Integrity Check
Ran 0.8% agarose 1x modified TAE gel stained with EtBr to assess the integrity of geoduck gDNA and Olympia oyster gDNA isolated yesterday.
Genomic DNA Isolation - Olympia oyster adductor musle & mantle
Previously isolated gDNA from these tissues on 20150901. However, found out after the isolations that BGI needs >73μg of gDNA for the genome sequencing project, which is significantly more than I obtained previously.
Genomic DNA Isolation - Geoduck Adductor Muscle & Foot
Previously isolated gDNA from these tissues on 20150828. However, found out after the isolations that BGI needs >73μg of gDNA for the genome sequencing project, which is significantly more than I obtained previously.
Agarose Gel - Geoduck & Olympia Oyster gDNA Integrity Check
Ran 0.8% agarose 1x modified TAE gel stained with EtBr to assess the integrity of [geoduck gDNA (from 20150828)(2015/08/28/genomic-dna-isolation-geoduck-adductor-muscle-foot.html) and Olympia oyster gDNA (from 20150901).
Genomic DNA Isolation - Olympia oyster adductor musle & mantle
Isolated gDNA from Ostrea lurida (Olympia oyster) adductor muscle & mantle samples collected by Brent & Steven on 20150812 using the E.Z.N.A Mollusc DNA Kit (Omega Bio-Tek) according to the manufacturer’s protocol, with the following adjustments:
Server Email Notifications Fix - Eagle
The system was previously set to use Steven’s Comcast SMTP server. Sending a test email from Eagle failed, indicating authentication failure. I changed this to use the University of Washington’s email server for outgoing messages. Here’s how…
Genomic DNA Isolation - Geoduck Adductor Muscle & Foot
Isolated gDNA from Panopea generosa (geoduck) adductor muscle & foot samples collected by Brent & Steven on 20150811 using the E.Z.N.A Mollusc DNA Kit (Omega Bio-Tek) according to the manufacturer’s protocol, with the following adjustments:
RAD-Seq Library Prep Reagents
A box with the above title was established in the -20C in FTR 209 containing the following:
SsoFast EvaGreen Supermix Aliquots
Prepared sterile, ~1.5mL aliquots of SsoFast EvaGreen Supermix (received 20150810) in 2.0mL screw cap tubes. All aliquots were dated and stored @ -20C in the “PCR Supplies” box.
Reverse Transcription – O.lurida DNased RNA 1hr post-mechanical stress
Performed reverse transcription on the Olympia oyster DNased RNA from the 1hr post-mechanical stress samples from Jake’s project. To accommodate the large numbers of anticipated genes to be targeted in subsequent qPCRs, I prepared 100μL reactions (normally, 25μL reactions are prepared) using 250ng of each DNased RNA. A 1:10 dilution of the oligo dT primers (Promega) was prepared to improve pipetting accuracy. All incubations were performed in a thermal cycler without using a heated lid.
qPCR - Jake’s O.lurida ctenidia 1hr post-mechanical stress DNased RNA
Ran qPCR on DNased RNA from earlier today to assess whether there was any residual gDNA after the DNase treatment with Oly_Actin_F/R primers (SR IDs: 1505, 1504).
RNA Quantification - O.lurida 1hr post-mechanical heat stress DNased RNA
DNased RNA from 07272015 was quantified using the Roberts Lab NanoDrop1000.
Goals - August 2015
Review of last month’s goals:
Sample Submission – Olympia oyster PCRs Sanger Sequencing
Submitted a plate of purified PCR products (PCR products prepared by Jake on 20150625) that Jake set up 20150625, to the UW High-Throughput Genomics Center for Sanger sequencing.
Server HDD Failure - Owl
We had our first true test of the Synology RAID redundancy with our Synology 1812+ server (Owl). One of the hard drives (HDD) failed. All of the other drives were fine, the data was intact and we had a new replacement HDD on hand. However, there was one shortcoming: no email notification of the drive failure. Luckily, the Synology server is next to Steven’s office and he could hear an audible beeping alerting him to the fact that something was wrong. In any case, the email notifications have been fixed and a replacement hard drive was added to the system. Here’s how these things were accomplished.
DNase Treatment - O.lurida Ctenidia 1hr Post-Mechanical Stress RNA
RNA Isolation – O.lurida Ctenidia 1hr Post-Mechanical Stress
Isolated RNA from [Jake’s Ostrea lurida ctenidia samples that had been subjected to mechanical stress (from 20150422)(https://heareresearch.blogspot.com/2015/04/4-22-2015-heatmechanical-shock.html).
RNA Isolation - O.lurida Ctenidia 1hr Post-Mechanical Stress
Isolated RNA from [Jake’s Ostrea lurida ctenidia samples that had been subjected to mechanical stress (from 20150422)(https://heareresearch.blogspot.com/2015/04/4-22-2015-heatmechanical-shock.html).
SsoFast EvaGreen Supermix Aliquots
Prepared sterile, 1.0mL aliquots of SsoFast EvaGreen Supermix (received today) in 2.0mL screw cap tubes. All aliquots were dated and stored @ -20C in the “PCR Supplies” box.
Automatic Notebook Backups - wget Script & Synology Task Scheduler
UPDATE 20150714 - READ ENTIRE POST
PCR - Sea Pen luciferase
Ran a PCR to obtain luciferase DNA for sequencing.
GOALS - July 2015
Before we check out this month’s goals, let’s have a quick review of last month’s goals and which, if any, I was able to accomplish:
Opticon2 Calibration
Jake and Steven recently noticed localized “hot spots” on most of Jake’s recent qPCRs, where higher levels of fluorescence were consistently showing up in interior portions of the plates than the outer portion of the plates.
RNAseq Data Receipt - Geoduck Gonad RNA 100bp PE Illumina
Received notification that the samples sent on 20150601 for RNAseq were completed.
Sample Submission – Olympia oyster PCRs Sanger Sequencing
Submitted a plate of purified PCR products (PCR products prepared by Jake on 20150623) that Jake set up yesterday, to the UW High-Throughput Genomics Center for Sanger sequencing.
Reverse Transcription - O.lurida DNased RNA Controls and 1hr Heat Shock
Performed reverse transcription on the Olympia oyster DNased RNA from the control samples and the 1hr heat shock samples of Jake’s project. To accommodate the large numbers of anticipated genes to be targeted in subsequent qPCRs, I prepared 100μL reactions (normally, 25μL reactions are prepared) using 250ng of each DNased RNA. A 1:10 dilution of the oligo dT primers (Promega) was prepared to improve pipetting accuracy. All incubations were performed in a thermal cycler without using a heated lid.
Sample Submission – Olympia oyster & Sea Pen PCRs Sanger Sequencing
Prepared two DNA plates and corresponding primer plates for sequencing at the UW HTGC from the purified gel-purified PCRs from yesterday. Primer plates were prepared by adding 7μL of NanoPure H2O to each well and then adding 3μL of 10μM primer to the appropriate wells. For the DNA plates, added 10μL of DNA to the appropriate wells.
Gel Purification - Olympia Oyster and Sea Pen PCRs
Purified DNA from the remaining PCR bands excised by Jake on 20150609 and 20150610, as well as Jonathan’s sea pen PCRs from 20150604, using Ultrafree-DA spin columns (Millipore). Transferred gel pieces from storage tubes (1.5mL snap cap tubes) to spin columns. Spun 10,000g, 5mins @ RT. Transferred purified DNA back to original storage tubes. See the sequence_log (Google Sheet) for a full list of the samples and the sequencing plates layouts. Purified DNA was stored @ 4C O/N. Will prepare and submit plates for Sanger sequencing tomorrow.
Sample Submission - Olympia oyster PCRs Sanger Sequencing
Submitted a plate of purified PCR products (PCR products prepared by Jake on 20150609 and 20150610) that Jake set up yesterday, to the UW High-Throughput Genomics Center for Sanger sequencing.
Sample Submission - Geoduck Gonad for RNA-seq
Prepared two pools of geoduck RNA for RNA-seq (Illumina HiSeq2500, 100bp, PE) with GENEWIZ, Inc.
Goals - June 2015
Before we check out this month’s goals, let’s have a quick review of last month’s goals and which, if any, I was able to accomplish.
Bioanalyzer - Geoduck Gonad RNA Quality Assessment
Before proceeding with transcriptomics for this project, we need to assess the integrity of the RNA via Bioanalyzer.
Reverse Transcription - Subset of Jake’s O.lurida DNased RNA
Currently don’t have sufficient reagents to perform reverse transcription on the entire set of DNased RNA (control and 1hr.heat-shocked O.lurida ctenidia samples). To enable Jake to start testing out some of his primers while we wait for reagents to come in, Steven suggested I generate some cDNA for him to use.
Bioinformatics – Trimmomatic/FASTQC on C.gigas Larvae OA NGS Data
Previously trimmed the first 39 bases of sequence from reads from the BS-Seq data in an attempt to improve our ability to map the reads back to the C.gigas genome. However, Mac (and Steven) noticed that the last ~10 bases of all the reads showed a steady increase in the %G, suggesting some sort of bias (maybe adaptor??):
qPCR - Re-run Jake’s O.lurida DNased RNA Samples NC1, SC1, SC2, SC4 from 20150514
The following DNased RNA samples showed inconsistencies between qPCR reps (one rep showed amplification, the other rep did not) on 20150514:
qPCR – Jake’s O.lurida ctenidia DNased RNA (1hr Heat Shock Samples)
Ran qPCR on DNased RNA from earlier today to assess whether there was any residual gDNA after the DNase treatment with Oly_Actin_F/R primers (SR IDs: 1505, 1504).
qPCR – Jake’s O.lurida ctenidia DNased RNA (Control Samples)
Ran qPCR on DNased RNA from earlier today to assess whether there was any residual gDNA after the DNase treatment with Oly_Actin_F/R primers (SR IDs: 1505, 1504).
DNase Treatment - Jake’s O.lurida Ctenidia RNA (1hr Heat Shock) from 20150506
Since the O.lurida RNA I isolated on 20150506 showed residual gDNA via qPCR, I treated 1.5μg of RNA from each sample using the Turbo DNA-free Kit (Ambion/Life Technologies), following the “rigorous” protocol.
DNase Treatment - Jake’s O.lurida Ctenidia RNA (Controls) from 20150507
Since the O.lurida RNA I isolated on 20150507 showed residual gDNA via qPCR, I treated 5μg of RNA from each sample using the Turbo DNA-free Kit (Ambion/Life Technologies), following the “rigorous” protocol.
ISO Creation - OpticonMonitor3 Disc Cloning
Since many newer computers are coming without optical disc drives (including my laptop, which I want to install this software on), I created an .iso disc image of the OpticonMonitor3 (BioRad) installation disc.
qPCR - Jake O.lurida ctenidia RNA (Heat Shock Samples) from 20150506
Ran qPCRs on the O.lurida total RNA I isolated on 20150506 to assess presence of gDNA carryover with Oly Actin primers (SR IDs: 1505, 1504).
qPCR - Jake O.lurida ctenidia RNA (Control Samples) From 20150507
Ran qPCRs on the O.lurida total RNA I isolated on 20150507 to assess presence of gDNA carryover with Oly Actin primers (SR IDs: 1505, 1504).
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
RNA Isolation – Jake’s O. lurida Ctenidia Control from 20150422
Isolated RNA from Jake’s Olympia oyster ctenidia, controls, collected on 20150422. Samples had been homogenized and stored @ -80C.
Bioinformatics - Trimmomatic/FASTQC on C.gigas Larvae OA NGS Data
In another troubleshooting attempt for this problematic BS-seq Illumina data, I’m going to use Trimmomatic to remove the first 39 bases of each read. This is due to the fact that even after the previous quality trimming with Trimmomatic, the first 39 bases still showed inconsistent quality:
RNA Isolation - Jake’s O. lurida Ctenidia 1hr Heat Stress from 20150422
Isolated RNA from Jake’s Olympia oyster ctenidia, 1hr heat shock, collected on 20150422. Samples had been homogenized and stored @ -80C.
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
BLAST - C.gigas Larvae OA Illumina Data Against GenBank nt DB
In an attempt to figure out what’s going on with the Illumina data we recently received for these samples, I BLASTed the 400ppm data set that had previously been de-novo assembled by Steven: EmmaBS400.fa.
Goals - May 2015
Here are the things I plan to tackle throughout the month of May:
BLASTN - C.gigas OA Larvae to C.gigas Ensembl 1.24 BLAST DB
In an attempt to figure out what’s going on with the Illumina data we recently received for these samples, I BLASTed the 400ppm data set that had previously been de-novo assembled by Steven: EmmaBS400.fa.
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
Bioanalyzer Data - Geoduck RNA from Histology Blocks
I received the Bioanalyzer data back for the geoduck foot RNA samples I submitted 20150422. The two samples were run on the RNA Pico chip assay.
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
Bioanalyzer Submission - Geoduck Gonad RNA from Histology Blocks
Submitted 3μL (~75ng) of RNA from each of the two gonad samples isolated from foot tissue embedded in paraffin histology blocks 20150408 (to assess quality of RNA) to Jesse Tsai at University of Washington Department of Environmental and Occupational Health Science Functional Genomics Laboratory:
Quality Trimming - C.gigas Larvae OA BS-Seq Data
Jupyter (IPython) Notebook: 20150414_C_gigas_Larvae_OA_Trimmomatic_FASTQC.ipynb
Quality Trimming - LSU C.virginica Oil Spill MBD BS-Seq Data
Jupyter (IPython) Notebook: 20150414_C_virginica_LSU_Oil_Spill_Trimmomatic_FASTQC.ipynb
Sequence Data Analysis - LSU C.virginica Oil Spill MBD BS-Seq Data
Performed some rudimentary data analysis on the new, demultiplexed data downloaded earlier today:
Sequence Data Analysis - C.gigas Larvae OA BS-Seq Data
Compared total amount of data generated from each index. The commands below send the output of the ‘ls -l’ command to awk. Awk sums the file sizes, found in the 5th field ($5) of the ‘ls -l’ command, then prints the sum, divided by 1024^3 to convert from bytes to gigabytes.
Sequence Data - C.gigas OA Larvae BS-Seq Demultiplexed
I had previously contacted Doug Turnbull at the Univ. of Oregon Genomics Core Facility for help demultiplexing this data, as it was initially returned to us as a single data set with “no index” (i.e. barcode) set for any of the libraries that were sequenced. As it turns out, when multiplexed libraries are sequenced using the Illumina platform, an index read step needs to be “enabled” on the machine for sequencing. Otherwise, the machine does not perform the index read step (since it wouldn’t be necessary for a single library). Surprisingly, the sample submission form for the Univ. of Oregon Genomics Core Facility doesn’t request any information regarding whether or not a submitted sample has been multiplexed. However, by default, they enable the index read step on all sequencing runs. I provided them with the barcodes and they demultiplexed them after the fact.
Sequence Data - LSU C.virginica Oil Spill MBD BS-Seq Demultiplexed
I had previously contacted Doug Turnbull at the Univ. of Oregon Genomics Core Facility for help demultiplexing this data, as it was initially returned to us as a single data set with “no index” (i.e. barcode) set for any of the libraries that were sequenced. As it turns out, when multiplexed libraries are sequenced using the Illumina platform, an index read step needs to be “enabled” on the machine for sequencing. Otherwise, the machine does not perform the index read step (since it wouldn’t be necessary for a single library). Surprisingly, the sample submission form for the Univ. of Oregon Genomics Core Facility doesn’t request any information regarding whether or not a submitted sample has been multiplexed. However, by default, they enable the index read step on all sequencing runs. I provided them with the barcodes and they demultiplexed them after the fact.
RNA Isolation - Geoduck Gonad in Paraffin Histology Blocks
Isolated RNA from geoduck gonad previously preserved with the PAXgene Tissue Fixative and Stabilizer and then embedded in paraffin blocks. See Grace’s notebook for full details on samples and preservation.
Sequencing Data - C.gigas Larvae OA
Our sequencing data (Illumina HiSeq2500, 100SE) for this project has completed by Univ. of Oregon Genomics Core Facility (order number 2212).
Epinext Adaptor 1 Counts - LSU C.virginica Oil Spill Samples
Before contacting the Univ. of Oregon facility for help with this sequence demultiplexing dilemma, I contacted Epigentek to find out what the other adaptor sequence that is used in the EpiNext Post-Bisulfite DNA Library Preparation Kit (Illumina). I used grep and fastx_barcode_splitter to determine how many reads (if any) contained this adaptor sequence. All analysis was performed in the embedded Jupyter (IPython) notebook embedded below.
TruSeq Adaptor Counts – LSU C.virginica Oil Spill Sequences
Initial analysis, comparing barcode identification methods, revealed the following info about demultiplexing on untrimmed sequences:
TruSeq Adaptor Identification Method Comparison - LSU C.virginica Oil Spill Sequences
We recently received Illumina HiSeq2500 data back from this project. Initially looking at the data, something seems off. Using FASTQC, the quality drops of drastically towards the last 20 bases of the reads. We also see a high degree of Illumina TruSeq adaptor/index sequences present in our data.
DNA Quantification - Claire’s C.gigas Sheared DNA
In an attempt to obtain the most accurate measurement of Claire’s sheared, heat shock mantle DNA, I quantified the samples using a third method: fluorescence.
Library Quality Assessment - C.gigas OA larvae Illumina libraries
Ran the 400ppm library and the 1000ppm library preps on a DNA1000 Assay Chip (Agilent) on the Agilent 2100 Bioanalyzer.
BS-seq Library Prep - C.gigas Larvae OA 1000ppm
Bisulfite Conversion
DNA Quantification - C.gigas Larvae 1000ppm
After the discovery that there wasn’t any DNA in the BS-seq Illumina library prep and no DNA in the bisulfite-treated DNA pool, I decided to try to recover any residual DNA left in the 1B2 sample. Sample 1B2 (sheared on 20150109) was dry, so I added 20μL of Buffer EB (Qiagen) to the tube. I vortexed both the 1B1 and 1B2 samples and quantified on the NanoDrop1000 (ThermoFisher). I also re-quantified the pooled BS-treated sample that had been used as input DNA for the libraries.
DNA Quantification - Claire’s Sheared C.gigas Mantle Heat Shock Samples
I previously checked Claire’s sheared DNA on the Bioanalyzer to verify the fragment size and to quantify the samples. Looking at her notebook, her numbers differ greatly from the Bioanalyzer, possibly due to the fact that the DNA1000 assay chip used only measures DNA fragments up to 1000bp in size. If her shearing was incomplete, then there would be DNA fragments larger than 1000bp that wouldn’t have been measured by the Bioanalyzer. So, I decided to quantify the samples on the NanoDrop1000 (ThermoFisher) again.
Bioanalyzer - C.gigas Sheared DNA from 20140108
To complement MBD ChiP-seq data and RNA-seq data that we have from this experiment, we want to generate, at a minimum, some BS-seq data from the same C.gigas individuals used for the other aspects of this experiment. Claire had previously isolated DNA and sheared the DNA on 20140108. If possible, we’d like to perform MBD enrichment, but the current quantities of DNA may prevent us from this.
Library Prep - Quantification of C.gigas larvae OA 1000ppm library
The completed BS Illumina library made on Friday (1000ppm) was quantified via fluorescence using the Quant-iT DNA BR Kit (Life Technologies/Invitrogen). Also quantified Jake’s libraries. Used 1μL of each sample and the standards. All standards were run in duplicate. Due to limited sample, the libraries were only processed singularly, without replication. Fluorescence was measured on a FLx800 plate reader (BioTek), using the Gen5 (BioTek) software for all calculations.
Sequencing Data - LSU C.virginica MBD BS-Seq
Our sequencing data (Illumina HiSeq2500, 100SE) for this project has completed by Univ. of Oregon Genomics Core Facility (order number 2112).
Bisulfite NGS Library Prep - Bisulfite Conversion & Illumina Library Construction of C.gigas larvae DNA
Bisulfite Conversion
Bisuflite NGS Library Prep – C.gigas larvae OA bisulfite library quantification
The two completed BS Illumina libraries (400ppm and 1000ppm) were quantified via fluorescence using the Quant-iT DNA BR Kit (Life Technologies/Invitrogen). Used 1uL of each sample and the standards. All standards were run in triplicate. Due to limited sample, the two libraries were only processed singularly, without replication. Fluorescence was measured on a FLx800 plate reader (BioTek).
Bisuflite NGS Library Prep - C.gigas larvae OA bisulfite DNA (continued from yesterday)
Continued Illumina library prep of bisulfite-treated DNA samples (400ppm and 1000ppm; from [20150114)(2015/01/14/dna-bisulfite-conversion-c-gigas-larvae-oa-sheared-dna.html) with Methylamp DNA Modification Kit (Epigentek). Performed bead clean up immediately after End Repair.
Bisuflite NGS Library Prep - C.gigas larvae OA bisulfite DNA
The two pooled bisulfite-treated DNA samples (400ppm and 1000ppm) from 20150114 were used to prepare bisulfite Illumina libraries with [EpiNext Post-Bisulfite DNA Library Preparation Kit (Illumina) (Epigentek)(https://github.com/sr320/LabDocs/blob/master/protocols/Commercial_Protocols/Epigentek_PostBisulfiteIlluminaLibraryPrep_P-1055.pdf). Changes to the manufacturer’s protocol:
Library Cleanup - LSU C.virginica MBD BS Library
I was contacted by the sequencing facility at the University of Oregon regarding a sample quality issue with our library. As evidenced by the electropherogram below, there is a great deal of adaptor primer dimer (the peak at 128bp):
DNA Bisulfite Conversion - C.gigas larvae OA Sheared DNA
After discussing with Steven, decided to use only samples from 20110513, due to high algae amounts present in the 20110519 samples.
SpeedVac - C.gigas larvae OA DNA
DNA Isolation - C.gigas larvae from 2011 NOAA OA Experiment
2014
Bisulfite NGS Library - LSU C.virginica Oil Spill MBD Bisulfite DNA Sequencing Submission
Combined the following libraries in equal quantities (17ng each) to create a single, multiplexed sample for sequencing (LSU_Oil_01):
Bisulfite NGS Library Prep - LSU C.virginica Oil Spill MBD Bisulfite DNA and Emma’s C.gigas Larvae OA Bisulfite DNA (continued from yesterday)
Continued library prep from yesterday. Set up Library Amplification according to the protocol. The samples received the following Barcode Indices:
Bisulfite NGS Library Prep - LSU C.virginica Oil Spill Bisulfite DNA and Emma’s C.gigas Larvae OA Bisulfite DNA
Constructed next generation libraries (Illumina) using the bisulfite-treated DNA from yesterday using the EpiNext Post-Bisulfite DNA Library Preparation Kit - Illumina (Epigentek). Samples were processed according to the manufacturer’s protocol up to Section 8 (Library Amplification) with the following changes:
Bisulfite Conversion - LSU C.virginica Oil Spill MBD DNA and Emma’s C.gigas Larvae OA DNA
DNA Isolation - Claire’s C.gigas Female Gonad for Illumina Bisulfite Sequencing
Due to poor “tag counts” from the initial sequencing (DATE) and the re-sequencing (20131127) of this sample, the HTGU facility has concluded that the library is probably at fault. They will make a new library and do a quality control run on the new library. However, they have insufficient gDNA left to make a new library.
EtOH Precipitation - LSU C.virginica Oil Spill MBD Continued (from 20141126)
Precipitation was continued according to the MethylMiner Methylated DNA Enrichment Kit (Invitrogen). Since I will need sample volumes of 24uL for the subsequent bisulfite conversion, I resuspended the samples in 29uL of water (will use 2.5uL x 2 reps for quantification).
Methylated DNA Enrichment (MBD) - LSU C.virginica Oil Spill gDNA
Enrichment was performed using the MethylMiner Methylated DNA Enrichment Kit (Invitrogen) according to the manufacturer’s protocol with the following changes:
Gel - Sheared LSU C.virginica Oil Spill gDNA (from yesterday)
Ran ~250ng of sheared C.virginica gDNA from yesterday’s shearing.
DNA Shearing - LSU C.virginica Oil Spill gDNA
Used the remainder of the “sheared” samples (see today’s earlier entry; ~2750ng). Brought the volumes up to 80uL and transferred to 0.5mL snap cap tubes. The volume of 80uL was selected because it’s above the minimum volume required for shearing in 0.5mL tubes (10uL according to the Biorupter 300 manual) and the MethylMiner Kit (Invitrogen) requires the input DNA volume to be <= 80uL.
Gel - Sheared gDNA
Ran ~250ng (out of 3000ng, according to Claire) of LSU C.virginica oil spill gDNA on a gel that was previously sheared by Claire to verify that shearing was successful.
RNA Seq - C.gigas Total RNA from Claire’s Pre/Post Heat Shock
Shipped (dry ice) ~15ug of total RNA from each of the following samples to Genewiz, Inc for high throughput transcriptomic sequencing (Illumina HiSeq2500, 100bp, single end).
DNA Isolation - C.gigas Larvae from Emma OA Experiments
Isolated gDNA using DNazol from the following larval samples for potential MBD selection and bisulfite sequencing:
RAD Sequencing - Oly Oyster gDNA-01 RAD Library (from 20141110)
Prepared 10nM of the library in total volume of 20uL of Buffer EB (Qiagen) + 0.1% Tween-20, according the University of Oregon’s Genomic’s Core:
DNA Quantification - Oly Oyster gDNA-01 RAD Library
Quantified the library using the Quant-It BR Kit (Life Technologies) according to the manufacturer’s protocol. Only used 1uL of the RAD library due to the small volume (15uL).
Library Prep - Oly Oyster gDNA-01 RAD
Used gel-purified, size-selected DNA from yesterday to prepare the RAD library using the Kappa LTP Kit:
DNA Shearing & Size Selection - Oly Oyster gDNA RAD P1 Adapters (from 20141105)
Pooled “low quality” samples and pooled “high quality” samples separately (in 1.5mL snap cap tubes) prior to shearing to improve chances of getting similar DNA size ranges.
Ligation - Illumina P1 Adapters for Oly Oyster gDNA-01 RAD Sequencing (from 20141031)
Made 25nM working stock from the 100nM stock adapter plate provided by Carita. Added 2uL of each adapter to corresponding well of SbfI digested DNA (e.g. DNA plate well A1 got the P1 adapter from well A1 in the adapter plate).
Restriction Digest - Oly Oyster gDNA-01 for RAD Sequencing (from 20141029)
Samples required two days of drying for all samples to fully dry down.
DNA Allocation - Oly Oyster gDNA-01 for RAD Sequencing (from 20141022)
Transferred 500ng of gDNA from each sample to a 96-well, low-profile, non-skirted PCR plate (sample layout matches that of the layout of the source gDNA plate) and air-dried the samples over night.
DNA Quantification - Oly Oyster gDNA-01 for RAD Sequencing (from 20141014)
Quantified the gDNA isolated 20141014 using the Quant-iT dsDNA Broad Range kit (Life Technologies).
DNA Isolation - Olympia Oyster Populations for RAD Sequencing
Olympia oysters from three different Puget Sound locations/populations (HL, NF, and SN) were collected and stored @ -80C on 8/29/2103.
Package - Received Package from Jerome LaPeyre from LSU
Oyster (C.virginica) gill samples exposed to “no oil” and “highest level of oil.” Samples were stored in Rack #2 in the -80C. Images of the box label and included paperwork below.
DNA Isolation - C.gigas Larvae from Katie Latterhos
Since the previous isolation attempt was unsuccessful (see 20140922), we’re trying a slightly different approach than yesterday.
DNA Isolation - C.gigas Larvae from Katie Latterhos and Emma
Isolated gDNA from two C.gigas larvae samples (stored in RNA Later) from Katie Latterhos:
PCR - Mac’s Bisulfite-Treated DNA
Realized that the PCR performed on 20140828 used the incorrect forward primer! As such, am repeating as before, but with the correct forward primer:
PCR - Mac’s Bisulfite-Treated DNA
Per Mac’s request, ran a PCR on a set of bisulfite-treated DNA (in her gDNA 2014 box in small -20C):
RNA-Seq - Sea Star Data Download
Received RNA-seq data from Cornell. They provided a convenient download script for retrieving all the data files at one time (a bash script containing a series of wget commands with each individual file’s URL), which is faster/easier than performing individual wget commands for each individual file and faster/easier then using the Synology “Download Station” app when so many URLs are involved.
RNA Isolation - Jessica’s Geoduck Larval Stages
Isolated RNA from the following samples provided by Jessica Blanchette (stored in RNA later):
DNA Isolation - Mackenzie’s C.gigas EE2 Gonad Samples
Isolated DNA from the following samples, provided by Mackenzie:
Sample Submission - Colleen Sea Star (Pycnopodia) Coelomycete RNA for Illumina Sequencing
Sent the following samples (in their entirety) to Cornell for Illumina HiSeq 100bp paired-end sequencing:
RNA Isolation - Colleen Sea Star (Pycnopodia) Coelomycete Sample
Apparently the Bio26 sample provided on 20140428 was incorrect. Instead, the sample should have been CF26.
RNA Isolation - Colleen Sea Star (Pycnopodia) Coelomycete Samples
Isolated RNA from the following samples (provided by Colleen Burge):
DNA Isolation - Claire’s C.gigas Female Gonad
Trying this sample again(!!), but will now use TE for pellet resuspension to prevent sample degradation. Incubated sample RT on rotator in 500uL of DNazol + 2.7uL of Proteinase K (Fermentas; Stock 18.5mg/mL) for 5hrs. Added additional 500uL of DNazol, mixed gently and followed DNazol manufacturer’s protocol. Performed first pellet was with 70% DNazol/ 30% EtOH solution. Resuspended pellet in 200uL of TE and spec’d on NanoDrop1000.
Ethanol Precipitation - Colleen’s Sea Star Coelomycete RNA from Yesterday
Performed an EtOH precipitation on the sea start RNA due to some residual column resin (?) in the tubes after elution.
RNA Clean Up - Colleen’s Sea Star Coelomycete RNA from 20140416
Zymoresearch support suggested putting the samples through another set of columns to help clean up the apparent phenol carryover that was seen (absorbance peak shifted to 270nm) in the initial isolation of these samples.
RNA Isolation - Colleens’ Sea Star Coelomycetes Samples
Isolated RNA from the following samples stored in RNAlater:
DNA Isolation - Test Sample
Due to the recent poor quality gDNA that has been isolated from C.gigas gonad, I decided to do a quick test using TE for DNA pellet resuspension in hopes that old Buffer EB (Qiagen) or old nuclease-free H2O (Promega) are to blame for the apparent, rapid degradation that I’ve experienced.
Phenol-Chloroform DNA Clean Up - Mac and Claire’s Samples (from 20140410)
Due to low 260/230 values and Mac’s smeary sample, performed a phenol-chloroform DNA cleanup on the samples isolated 20140410.
RNA Isolation - Sea Star Coelomocytes (from Colleen)
Isolated RNA from two samples stored in RNAlater that had either no visible pellet or a minutely visible pellet:
DNA Isolation - Claire’s C.gigas Female Gonad and Mac’s C.gigas Gonad
Due to the poor quality DNA yielded by the DNeasy Kit (Qiagen; see 20140404), I am re-isolating these samples using DNazol (Molecular Research Center). Weighed tissue from each frozen sample:
Cloned Hard Drive - Windows XP Opticon Computer (Aquacul8)
Cloned the XP hard drive of the computer hooked up to the Opticon qPCR machine using Macrium Reflect Free. Verified that the clone is bootable and operates correctly. Will store one of the hard drives.
DNA gel - Claire’s C.gigas Female Gonad and Mac’s C.gigas Gonad
Ran out 2uL of Clair’es C.gigas female gonad gDNA (from 20140328) and Mac’s C.gigas gonad gDNA (from 20140402) for quality assessment. Both samples had been isolated using Qiagen’s Blood & Tissue DNeasy Kit. 2uL of each sample was run on a 0.8% 1x TBE gel.
RNA Isolation - Sea Star Coelomocytes (provided by Colleen Burge)
Tried another method of RNA Isolation for comparison with regular TriReagent method.
RNA Isolation - Sea Star Coelomocytes (provided by Colleen Burge)
Isolated RNA from the following samples (stored in RNAlater):
DNA Isolation - Mackenzie’s C.gigas Gonad Sample
Mac’s been having some difficulty getting good quality gDNA from some of her gonad samples, so she asked me to give it a shot.
DNA Isolation - C.gigas Female Gonads (from frozen)
Isolated gDNA from Claire’s “Female DNA” (from 05/16/2013) using the Qiagen Blood & Tissue DNeasy Kit according to the manufacturer’s protocol, with the following changes:
DNA Quality Check - Yanouk’s Oyster gDNA
We’ve had some Illumina sequencing issues with Yanouk’s samples, so I ran the samples out on a 0.8% agarose gel to evaluate the levels of degradation. Loaded 2uL of each sample. Did not load equal quantities of gDNA, due to the lack of available gDNA in the samples we submitted for Illumina sequencing. Added 2uL of H2O to samples 37 & 38 in hopes of having sufficient DNA for visualization on the gel.
DNA Isolation - Geoduck
Isolated additional geoduck gDNA from the two fresh (now frozen) geoduck’s that Brent provided me with on 20140212 so that we can potentially isolate RNA from the same geoducks to tie in with the DNA Illumina sequencing that Axa will be conducting. Isolated DNA using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol (incubated minced siphon tissue at 56C for 3hrs). Eluted with 75uL of ddH2O and spec’d on NanoDrop1000.
DNA Precipitation - Geoduck DNA from 20140213
After speaking with Axa regarding the DNA concentrations, he would like the DNA from the ethanol-fixed tissue to be more concentrated, and he wants them in ddH2O instead of Buffer AE (from the Qiagen DNeasy Kit). So, I preformed a standard ethanol precipitation. Added 0.1 volumes of 3M sodium acetate (pH = 5.2) [15uL], 2.5 volumes of 100% ethanol [412.5uL] and incubated @ -20C over the weekend.
DNA Isolation - Geoduck
Since yesterday’s DNA isolation failed to yield sufficient quantity of DNA from the ethanol-fixed samples, I isolated additional DNA from the same samples.
DNA Isolation - Geoduck
Isolated gDNA from 6 geoduck siphon samples provided by Brent using the Qiagen DNeasy Spin Kit. Samples were as follows:
2013
Gylcogen Assay - Emma’s C.gigas Whole Body Samples (continued from yesterday)
Samples from yesterday were centrifuged 30mins, 4000g, 4C (fixed angle rotor).
Gylcogen Assay - Emma’s C.gigas Whole Body Samples
Finally located the remaining half of Emma’s samples. These had already been freeze dried AND pulverized! So, I just had to weigh out ~half of each sample for the glycogen assay.
Gylcogen & Carboyhydrate Assays - Emma’s C.gigas Whole Body Samples (continued from yesterday)
Samples from yesterday were centrifuged 30mins, 4000g, 4C (fixed angle rotor).
Gylcogen and Carboyhydrate Assays - Emma’s C.gigas Whole Body Samples
Samples were previously freeze dried overnight and stored @ -20C. To maximize sample homogeneity and, thus, increase accuracy of both assays, all samples were mechanically pulverized in their existing tubes. Approximately half of each sample was weighed and used for the glycogen assay. The remainder of each sample was stored @ -20C.
PCR - Lake Trout C1q
PCR - Lake Trout C1q
DNA Quantification - Yanouk’s DNA Samples
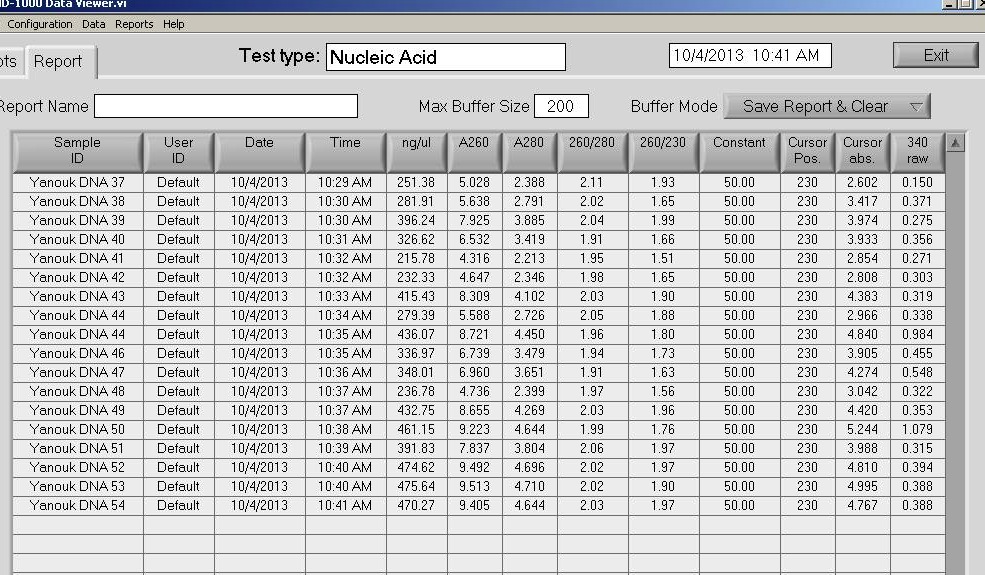
PCR - Lake Trout C1q
Ran PCR on lake trout DNA and lake trout bisulfite-converted DNA. Used primers SRIDs: 1551 and 1552. DNA was isolated by Caroline Storer on 4/4/2011 and bisulfite converted on 4/7/2011. Master mix and cycling params are here:
PCR - Hexokinase Partial CDS
Performed PCR using the primers CG_HK_CDS_2132-2158 (SRID: 1521) and Cg_Hk_CDS_3’_no_stop (SRID: 1519) on pooled C.gigas cDNA (from DATE).
PCR - Hexokinase and Partial Exon #1
Performed PCR using newly designed primers to amplify the C. gigas hexokinase “promoter” (-2059bp from start) along with a portion of the first exon.
PCR - Hexokinase Promoter and CDS (repeat from 20130227)
Performed a repeat of the failed PCR from 20130227, but used a pool of cDNA (made from 20110311 C.gigas cDNA) instead of a single sample and changed the annealing temp to 50C.
PCR - Hexokinase Promoter and CDS
Performed PCR to amplify the C.gigas hexokinase (ACCESSION#) promoter region (-2059bp) and the CDS without the stop codon. Elimination of the stop codon allows for subsequent cloning into the pBAD-TOPO expression vector, which will incorporate the V5 epitope tag sequence. This tag will be used to distinguish between endogenous hexokinase expression and expression generated from our hexokinase construct.
Reverse Transcription - Herring RNA from 20091026
Performed an RT reaction on pooled herring gonad and liver mRNA from 20091026 for James Raymond at the UNLV. A single RT reaction was performed using 12.75uL (208ng) of the pooled gonad mRNA and 5uL (132.5ng) of the pooled liver RNA, according to our default MMLV (Promega) protocol. After reaction was completed, sample was stored @ -20C and then shipped to James Raymond on 20130214.
2012
qPCR - Halley cDNA Check
Ran qPCR on Halley’s cDNA to see if I could get them to work. She has been getting high levels of fluorescence at the initiation of the qPCR cycling that shouldn’t be there. Master mix calcs and plate layout can be seen here. http://eagle.fish.washington.edu/Arabidopsis/Notebook%20Workup%20Files/20121128%20qPCR%20Layout.jpg
Reverse Transcription - FISH441 RNA
Reverse transcribed the class FISH441 RNA to cDNA. Followed protocol provided on the FISH441 Wiki page, NOT the usual Roberts Lab protocol, with some modifications.
qPCR - Manila Clam Larvae cDNA (from August 2012 - Dave’s Notebook)
Ran qPCR on manila clam larvae cDNA that Dave made on 8/7/2012, using the sample sets from 7/29/2011 and 8/5/2011 of the OA manila clam experiment he ran.
qPCR - Manila Clam Larvae cDNA (from August 2012 - Dave’s Notebook)
Ran qPCR on manila clam larvae cDNA that Dave made on 8/7/2012, using the sample sets from 7/29/2011 and 8/5/2011 of the OA manila clam experiment he ran.
qPCR - Manila Clam Larvae cDNA (from August 2012 - Dave’s Notebook)
Ran qPCR on manila clam larvae cDNA that Dave made on 8/7/2012, using the sample sets from 7/29/2011 and 8/5/2011 of the OA manila clam experiment he ran.
qPCR - DNased Manila Clam Larvae RNA (from August 2012 - Dave’s Notebook)
Performed qPCR on Dave’s manila clam larvae DNased RNA from August 2012 using EF1a primers (SR IDs: 1463, 1474).
Received oysters from Taylor Shellfish.
Photos of oysters are here:
qPCR - Opticon Test
Ran qPCR to test uniformity of Opticon 2, after it was serviced on 20120926.
Minipreps - Emma’s Illumina Library Cloning
Performed plasmid isolation on 17 bacterial cultures Emma inoculated yesterday using the QIAprep Spin Mini Kit (Qiagen) using ~1.4mL of culture according to the manufacturer’s protocol. Plasmid DNA was eluted with 50uL of Buffer EB. Tubes were stored @ 4C in the refrigerator in FTR 213.
Illumina RNAseq Library Construction - 32 C.gigas Individuals
Took heat-fragmented RNA provided by Emma (see Emma’s Notebook, 7/3/2011) and proceeded to make first strand cDNA, as described in the Eli Meyer protocol for Illumina HiSeq. Master mix calcs are here. Samples were stored @ -20C after the reverse transcription and library construction will be continued tomorrow.
Oligo Reconstitution - Illumina RNAseq Library Oligos and Barcodes
Reconstituted all of the oligos and barcodes for library construction in TE (pH = 8.0) to a final concentration of 100uM. Created 10uM working stocks of all oligos and barcodes. All samples (stocks and working stocks) are stored @ -80C in their own box (Illumina Library Oligos & Barcodes) due to the fact that one of the oligos is an RNA oligo and requires storage at -80C.
Chloroform Clean Up - Lexie’s QPX RNA from 20110504
After submission of QPX samples to HTGU for Illumina library prep yesterday, I was notified that there was insufficient RNA for the QPX RNA samples. I checked the source RNA on the Roberts Lab NanoDrop1000 and determined that they had high phenol contamination (large peak at 270nm), which results in a large exaggeration in the OD260 absorbance (NanoDrop1000 report[JPEG]; notice terrible OD260/280 ratios; did not save screen shot of absorbance peaks.). As such, the concentrations that Lexie had listed in her notebook for these samples are highly inaccurate and highly inflated. To remove the phenol, I brought all of her QPX RNA samples from 20110504 up to ~200uL with 0.1%DEPC-H2O, added 200uL of chloroform, vortexed for 30s, spun at 12,500g RT for 15mins, and transferred aqueous phase to new tube. Then performed an ethanol precipitation on the aqueous phase. Added 0.1 vols of 3.0M sodium acetate (pH = 5.2), 2.5 vols of 100% EtOH, mixed and incubated at -20C for 1hr. Pelleted RNA by spinning at 16,000g 4C for 15mins.
Sample Submission - QPX RNA and DNA for Illumina 36bp single-end RNA/DNAseq
Submitted samples to HTGU for Illumina sequencing.
QPX Sample Pooling for Illumina Sequencing
Pooling volumes/quantities for QPX RNA/DNA-seq: 20120705 Oly larvae and QPX HiSeq Calcs
qPCR - Detection of V.tubiashii Presence and Expression Using VtpA Primers in DNA/cDNA from yesterday
Ran qPCR with VtpA primers on cDNA and DNA (from yesterday) of C.gigas larvae to see levels of V.tubiashii compared to their water filter samples (see 20120326). Master mix calcs are here. Plate layout, cycling params, etc can be seen in the qPCR Report (see Results). Used 1uL of cDNA and 100ng (1uL) of DNA as template.
Reverse Transcription - DNased C.gigas Larval RNA from 20120427
Performed reverse transcription using random primers (Promega) diluted 1:100 (5ng/uL) with 175ng of DNased total RNA. Random primers were used because we will be targeting V.tubiashii RNA instead of eukaryotic RNA. Reverse transcription was performed with M-MLV Reverse Transcriptase (Promega) according to the manufacturer’s protocol. Calcs are here.
qPCR - Check DNased RNA from Earlier Today for Residual gDNA
Ran qPCR using V.tubiashii VtpA primers (from Elene; no SR ID). Used 0.5uL of each DNased RNA sample, which equals ~40ng of RNA, which would be the equivalent amount of RNA that would end up in a qPCR rxn after cDNA has been made (using 1uL of cDNA). Used the filter DNA extraction from samples #279 from DATE as a positive control. Master mix calcs are here. Plate layout, cycling params, etc. can be found in the qPCR Report (see Results).
DNase Treatment - C.gigas Larvae RNA from yesterday
Treated 5ug of total RNA (in a 50uL rxn) using Turbo DNA-free (Ambion) according to the “Standard” protocol. Samples were spec’d on the Roberts Lab NanoDrop 1000.
gDNA Isolation - C.gigas Larvae from Taylor Summer 2011
Samples that had been split from earlier today (see the RNA Isolation below) were resuspended in 1mL of DNAzol (MRC). 100ug of Proteinase K (Fermentas) was added to each sample. Samples were incubated at RT, O/N on a rotator. On 20120427 samples were pelleted 10mins, 10,000g, and supe transferred to fresh tube. DNA was precipitated with 0.5mL of 100% EtOH, mixed gently and pelleted 5mins, 5000g. Supe was discarded, pellets were washed with 1mL 75% EtOH, re-pelleted at same speed as previous step, supe discarded and pellets were resuspended in NanoPure H2O. Samples were spec’d on the Roberts Lab NanoDrop1000.
RNA Isolation - C.gigas Larvae from Taylor Summer 2011
Samples had been stored in RNA Later (Ambion). Samples were pelleted and the RNA Later supe removed. Samples were washed (2x) with 1mL TE (pH = 8.0) to remove excess salt resulting from the RNA Later. Samples were split, roughly equally, into two separate tubes. Samples were pelleted and the supe removed. One tube from each sample was set aside for gDNA isolation using DNAzol (MRC). The other tube was vortexed vigorously in TriReagent (MRC) and the then treated according to protocol. Samples were resuspended in 100uL of 0.1% DEPC-H2O and spec’d on the Roberts Lab NanoDrop 1000.
qPCR - Taylor Water Filter DNA Extracts from 20120322
Ran qPCR on the Taylor water filter DNA extracts from 20120322 using V.tubiashii VtpA primers (provide by Elene; no SR ID?) instead of 16s primers, which failed to produce acceptable results in the melt curves (see 20120323). Additionally, Elene has a standard curve for V. tubiashii (from 1/12/2011) based off of CFUs/mL, which will allow us to quantify theoretical number of V.tubiashii CFUs present in each sample.
qPCR - Repeat of qPCR from Earlier Today
Repeated exactly what was done earlier today due to apparent contamination in negative controls.
qPCR - Taylor Water Filter DNA Extracts from Yesterday
Ran qPCR on the Taylor water filter DNA extracts from yesterday using V.tubiashii 16s primers (SR IDs: 455, 456). Used RE22 DNA as a positive control, provided by Elene. Master mix calcs are here. All samples were run in duplicate. Plate layout, cycling params, etc can be found in the qPCR Report (see Results).
DNA Extraction - Taylor Water Filter Samples from 2011
Extracted DNA from the following water filter samples using the Qiagen DNeasy Blood & Tissue Kit:
Reverse Transcription - Dave’s Manila Clam (Venerupis philippinarum) DNased RNA from 20120307 and 20120302
Performed reverse transcription on 1.5ug of DNased RNA in a 75uL reaction, using oligo dT primers. All reagents were scaled appropriately (based on Promega’s M-MLV RT protocol). Samples were prepared in a plate and stored @ -20C. Plate layout and all reverse transcription calcs are here:
qPCR - Dave’s Manila Calm (Venerupis philippinarum) DNased RNA from yesterday and 20120302
Performed qPCR on all DNased RNA samples from this group (samples #1-48) using beta actin primers (SR IDs: 1379, 1380). 0.5uL of each DNased RNA was used, which was the equivalent of ~40ng, in order to simulate the amount of RNA present in the subsequent cDNA (1000ng of RNA in 25uL cDNA; use 1uL of cDNA in qPCR reaction). Master mix calcs are here. Plate layout, cycling params, etc., can be found in the qPCR Report (see Results). 0.5uL of total RNA from sample Vp gill 01 was used to serve as a positive control, since Dave has no existing V. phillippinarum cDNA.
RNA Isolation - Dave’s Manila Clam (Venerupis philippinarum) Gill Samples (#25-48)
Isolated RNA from Manila Clam gill samples provided by Dave, according to protocol. Samples were resuspended in 0.1%-DEPC H2O and spec’d on the Roberts Lab NanoDrop1000. Samples were stored @ -80C in Dave’s box that the tissue was initially stored in.
DNase - Dave’s Manila Clam (Venerupis philippinarum) Gill RNA from Yesterday
DNased RNA using Ambion’s Turbo DNA-free Kit following the “routine” protocol. 5ug of total RNA from each sample was treated in 50uL reactions. Samples will be spec’d on Monday with the Roberts Lab NanoDrop 1000.
RNA Isolation - Dave’s Manila Clam (Venerupis philippinarum) Gill Samples (#1-24)
Isolated RNA from Manila Clam gill samples provided by Dave according to protocol. Samples were resuspended in 0.1%-DEPC H2O and spec’d on the Roberts Lab NanoDrop1000. Samples were stored @ -80C in Dave’s box that the tissue was initially stored in.
qPCR - cDNA from 20120208
Performed qPCR on all 12 samples. Used the following primers, provided by Elene, to detect V.tubiashii expression:
qPCR - cDNA from earlier today
Performed qPCR on all 12 samples. Used Cg_EF1aF/R2 (SR IDs: 1410 & 1412) for one set of qPCRs and Vtub_16s_F/R (SR IDs: 455 & 456) for the other set of qPCRs. Used pooled C.gigas cDNA (from 20110311) and RE22 DNA (provided by Elene) as positive controls for C.gigas and V.tubiashii, respectively. C.gigas gDNA (7ng of BB16 from 20110201) was used as a negative control for EF1a. Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results). All samples were run in duplicate.
Reverse Transcription - C.gigas larvae DNased RNA (from 20120125)
Performed reverse transcription on Dnased RNA from 20120125 using 175ng RNA from each sample. Also used random primers (instead of the usual Oligo dT primers) since these samples will also be used to analyze gene expression in Vibrio tubiashii. cDNA calcs are here.
qPCR - DNased RNA from earlier today
Checked DNased RNA samples from earlier today for the presence of residual gDNA. Used C.gigas BB16 gDNA (from 20110201) diluted to ~7ng/uL as a positive control to match the dilution factor of the RNA that will be used in the reverse transcription reaction (175ng in 25uL = 7ng/uL). All samples were run in duplicate. Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results).
DNAse - C.gigas RNA from 20120124
5ug of each RNA was DNased using Ambion’s Turbo DNA-free Kit, according to the rigorous protocol and spec’d on Roberts Lab NanoDrop 1000. RNA volume calcs are here.
RNA Isolation - C.gigas Larvae from 20110412 & 20110705 (Continued from 20120112)
All of the RNA samples were re-combined with their respective counterparts and subject to a standard EtOH precipitation (0.1 volumes of 3M NaOAc, pH = 5.2, 2.5 volumes 100% EtOH; incubated -80C 1hr; pelleted; washed with 1mL 70% EtOH; pelleted). Pellets were washed two additional times (for a total of three washes) with 70% EtOH. RNA was resuspended in 50uL of 0.1% DEPC-H2O and spec’d on the Roberts Lab NanoDrop 1000.
RNA Isolation - C.gigas Larvae from 20110412 & 20110705
RNA was isolated from C.gigas larvae collected from Taylor Shellfish hatchery on the dates noted above. Samples were in RNA Later. RNA Later was removed. Attempted homogenization with a pestle proved futile, as a significant quantity of larvae were sticking to the pestle and were nearly impossible to wash off using TriReagent as a rinsing agent. Due to this, all samples were vortexed for 1min in 1mL of TriReagent. It should be noted that the TriReagent took on a cloudy appearance and even showed some separation into two layers upon letting the samples sit. This was not normal and I was immediately concerned about the high salt content from residual RNA Later. Samples were treated normally with the following changes:
2011
Sequencing - COX/PGS Clones from yesterday/today
Samples were submitted for sequencing to the University of Washington HTGU, two times from each direction using vector M13F/R primers. See Sequence Log for plate layout.
Mini-preps - COX/PGS Cloning Colonies from today
Selected 10 colonies (1-8, 18, 28) for mini-preps. Inoculated 5mL 1x LB + 50ug/mL of Kanamycin. Incubated O/N, 37C, 200RPM. 3mL of each culture were used for mini-preps. Used Qiagen kit. Samples were eluted w/30uL of EB.
PCR - COX/PGS Cloning Colony Screens from yesterday
Performed PCR on 40 colonies using both qPCR primer sets to see if I could differentiate between which colonies potentially contained each isoform to reduce the amount of clones needed for sequencing. Master mix and cycling params are here. Primers used were:
Cloning - Purified COX/PGS “qPCR Fragment” from 20111006
Cloned the purified “qPCR Fragment” from 20111006 using the TOPO TA Cloning Kit (Invitrogen). Performed a half reaction (total volume = 3uL), using 1uL of purified PCR product. Incubated at RT for 20mins and then placed reaction on ice. Transformed chemically competent TOP 10 cells (Invitrogen) and heat shocked at 42C for 30s. Added 250uL of RT S.O.C. medium and incubated at 37C, 200RPM. Plated cells on pre-warmed Kan50+X-Gal plates (plates from 20110726; X-Gal added ~30mins before plating cells). Incubated plates O/N, 37C.
PCR - Purified COX/PGS 1/2 DNA from earlier today
Ran PCR using primers Cg_COX1/2_qPCR_F, Cg_COX1_qPCR_R, Cg_COX2_454align1_R (SR IDs: 1192, 1191, 1190; respectively). Template was pooled cDNA from 20110311 of various C.gigas tissues. These reactions will verify (sort of) if we have both isoforms present in the PCR performed earlier today, prior to cloning. Master mix calcs and cycling params are here.
PCR - Region Outside of COX/PGS qPCR Primers
Ran PCR using primers Cg_COX_982_F and Cg_COX_2138_R (SR IDs: 1149 & 1151, respectively). Template was pooled cDNA from 20110311 of various C.gigas tissues. These primers anneal 5’ and 3’ of where the qPCR primers for both COX1/PGS1 and COX2/PGS2 anneal. Master mix calcs and cycling params are here. Ran multiples of the same reaction to ensure sufficient product for use in cloning/PCR.
Ethanol Precipitation - Full-length PGS1 cDNA (from 20110921)
Performed an EtOH on gel-purified PCR products from 20110921. Briefly, added 0.1 vols of 3M sodium acetate (pH=5.2; 43uL), mixed and then added 2.5 vols of 100% EtOH (1182.5uL). Mixed, split into two tubes (due to high volume not fitting in a single tube) and incubated @ -80C O/N. Pelleted DNA 16,000g, 20mins, 4C. Discarded supe. Washed pellet w/ 1mL 70% EtOH. Pelleted DNA 16,000g, 15mins, 4C. Discarded supe. Resuspended both pellets in a TOTAL of 25uL Qiagen Buffer EB (10mM Tris-HCl) and spec’d.
PCR - Full-length PGS1 cDNA
Still have insufficient quantities of DNA for sequencing. Master mix calcs and cycling params are here. Additionally, used some of the purified PCR product as template in one of the reactions, just for comparison purposes. cDNA template was pooled cDNA from 20110311 from various C.gigas tissues. Also, increased the amount of template 4-fold in an attempt to obtain higher yields of PCR products for sequencing.
Ethanol Precipitation - Purified PGS1 PCR from yesterday
Added 0.1 vols of 3M sodium acetate (pH = 5.2; 38uL) and 2 vols of 100% EtOH (836uL). Incubated 30mins @ -20C. Pelleted DNA 16,000g, 15mins, 4C. Removed supe. Washed pellet w/1mL 70% EtOH. Pelleted DNA 16,000g, 15mins, 4C. Removed supe. Air-dried pellet. Resuspended in 50uL of Qiagen EB Buffer and spec’d.
PCR - Full-length PGS1 cDNA
Need more PCR product for sequencing. Repeated reaction from 20110825.
PCR - Full-length PGS2 cDNA
Repeated PCR from 20110825 to attempt to amplify the full-length cDNA for PGS2 (COX2), however this time using a more robust polymerase (Amplitaq Gold) in hopes of getting results. Additionally, tried 3 different Mg2+ concentrations (1.5mM, 2.0mM, and 3.0mM). Master mix calcs and cycling params are here. cDNA was pooled cDNA made 20110311 from various tissues. PGS2 primers = 1376, 1375.
PCR - Full-length PGS1 & PGS2 cDNAs
Ran PCR to amplify full-length cDNAs of PGS1 & PGS2 (COX1 & COX2) using primers designed to anneal in the 5’/3’UTRs of each isoform. PGS1 primers = SRIDs: 1377, 1378. PGS2 primers = 1376, 1375. Master mix calcs and cycling params are here. cDNA was pooled cDNA made 20110311 from various tissues.
qPCR - C.gigas V.vulnificus Exposure cDNA (from 20110311)
Ran a qPCR using 3hr Vibrio vulnificus exposure cDNA from 20110311. Original experiment conducted on 20110111 with defensin primers (SR IDs: 1109 & 1070) and GAPDH (SR IDs: 1172 & 1173). Master mix calcs are here. Cycling params, plate layout, etc can be seen in the qPCR Report (see Results). This was performed to help Herschel.
Sequencing - C.gigas COX2/PGS2 Clone #4 from 20110728
Used new primers for sequencing (SR IDs: 1351 & 1352) clone #4. Sequenced clone two times in each direction. DNA and primers were sent for sequencing at ASU. Requested “High GC” treatment to help overcome the issue seen on 20110728.
Plasmid Isolation & Sequencing - C.gigas COX2/PGS2 Clones (from yesterday)
Isolated plasmid DNA from 3mL of liquid cultures that were inoculated yesterday using Qiagen’s miniprep kit. DNA was eluted with 50uL of EB. DNA was prepped and sent for sequencing to ASU sequencing facility. Each clone was sequenced two times in each direction. Samples are as follows:
Bacterial Cultures - C.gigas COX2/PGS2 Clones (from yesterday)
Inoculated 4 x 5mL 1xLB + Kan50 with a colony from each set of clones, incubated 37C, 200RPM, O/N. Will mini prep and send for sequencing tomorrow.
Colony PCRs - C.gigas COX2/PGS2 Clones (from yesterday)
Performed colony PCRs on the 4 sets of cloning reactions that were performed yesterday using the M13F/R vector primers. Colonies were picked, restreaked on a fresh LB Kan50 plates (made 20110726 by SJW) and PCR’d. Master mix calcs are here. Selected 8 white colonies from each cloning reaction for PCR. Restreaked plate was incubated @ 37C O/N.
Cloning - C.gigas COX2/PGS2 5’/3’ RACE Products (from earlier today)
The bands that were excised and purified earlier today were cloned in to pCR2.1 using the TOPO TA Cloning Kit (Invitrogen) according to the manufacturer’s protocol with the following changes: used 4uL of all PCR products, incubated ligation reactions for 15mins @ RT, incubated competent cells with ligation reactions for 15mins on ice.
5’/3’ RACE - C.gigas COX2/PGS2 Nested RACE PCR
Performed nested RACE PCR on the RACE PCR products generated on 20110722 using the following nested primers: PGS2_ngspRACE_5’ (SR ID: 1350) and PGS2_ngspRACE_3’ (SR ID: 1349). Removed 2uL from each of the primary PCR reactions and brought up to 100uL in tricine EDTA (supplied in the Clontech SMARTer RACE cDNA Amplification Kit). Performed the nested RACE PCR according to the Clontech manual. Briefly, this is the same as the primary RACE PCR reaction, but using 5uL of the diluted primary PCR product and 1uL of the Nested Universal Primer (instead of 5uL of the 10X Universal Primer Mix). Master mix calcs and set up are here. Cycling params followed “Program 2” of the Clontech protocol, modified for nested primers, and are as follows:
5’/3’ RACE - C.gigas COX2/PGS2 RACE PCR
Additional RACE using gene specific primers (SR IDs: 1347 & 1348) for C.gigas COX2/PGS2 according to Clontech’s SMARTer cDNA RACE Kit protocol. 3’/5’ RACE cDNA libraries are from 20080619. Master mix calcs and set up is here. Cycling params followed “Program 2” of the Clontech protocol and are as follows:
Sequencing - PGS Hi 4 (PGS2/COX2)
Sent plasmid prep to ASU (5uL of plasmid + 1uL of 10uM M13F/R). SJW01 = M13F, SJW02 = M13R.
Plasmid Isolation - Miniprep on PGS Hi 4 Colony from yesterday
Plasmid was isolated from 3mL of liquid culture started yesterday using the Qiagen MiniPrep Kit, according to the manufacturer’s protocol. Will send off for sequencing.
PCR - Colony PCR on Restreaked PGS2 Clones from 20110707
Ran a colony PCR at the same time that I inoculated liquid cultures, using M13 primers.
Bacterial Cultures - Liquid Cultures of PGS2/COX2 Colonies from 20110707
Clone Restreaking - PGS2 Hi/Lo Clones (from 20110421)
Sequencing of the PGS2/COX2 clone failed (was empty vector). Restreaked bacterial clones on to a Kan50 plate (made 20110413 by SJW) from a plate that Caroline Storer had created from cloning colony selection on 20110421. Samples were labeled as PGS Lo 1 & 2 and PGS Hi 3 & 4. Additionally, there were red numbers on the plate associated with these four samples. They were 42 - 45, respectively. PGS Hi 4 (#45) was previously grown up and sequenced. This sample is what produced vector only sequence. Incubated O/N @ 37C. Hopefully we’ll bacteria is still viable and will have samples to grow up for miniprep, plamsid iso and sequencing.
qPCR - C.gigas GAPDH second rep on V.vulnificus exposure cDNA (from 20110311) and standard curves for COX1, COX2, GAPDH
Ran a qPCR on all cDNA samples. Created a standard curve to possibly allow for use of the BioRad software for gene expression analysis. Standard curve was created from pooled cDNA (1uL from each individual sample). Master mix calcs are here.
qPCR - C.gigas actin and GAPDH on V.vulnificus exposure cDNA (from 20110311)
Ran a qPCR on all cDNA samples from the V.vulnificus exposure experiment from 20110111. This qPCR was to test 2 of 4 potential normalizing genes to evaluate which genes show the least amount of effect from the treatments in this experiment. Primers for actin used were Cg_Actin_306_F (SR ID: 1170), Cg_Actin_408_R (SR ID: 1171). Samples were run in duplicate. Master mix calcs are here. The master mix info is the same that was used earlier today, but with the primers noted above, not those listed on the calcs page. Plate layout, cycling params, etc., can be seen in the qPCR Report (see Results).
qPCR - C.gigas 18s and EF1a on V.vulnificus exposure cDNA (from 20110311)
Ran a qPCR on all cDNA samples from the V.vulnificus exposure experiment from 20110111. This qPCR was to test 2 of 4 potential normalizing genes to evaluate which genes show the least amount of effect from the treatments in this experiment. Primers for 18s used were Cg_18s_1644_F (SR ID: 1168), Cg_18s_1750_R (SR ID: 1169). Primers for EF1a used were EF1_qPCR_5’ (SR ID: 309), EF1_qPCR_3’ (SR ID: 310)Samples were run in duplicate. Master mix calcs are here. The master mix info is the same that was used earlier today, but with the primers noted above, not those listed on the calcs page. Plate layout, cycling params, etc., can be seen in the qPCR Report (see Results).
qPCR - C.gigas COX2 on V.vulnificus exposure cDNA (from 20110311)
Ran a qPCR on all cDNA samples from the V.vulnificus exposure experiment from 20110111. Primers used were Cg_COX1/2_qPCR_F (SR ID: 1192) & Cg_COX2_454align1_R (SR ID: 1190). Samples were run in duplicate. Master mix calcs are here. The master mix info is the same that was used earlier today, but with the primers noted above, not those listed on the calcs page. Plate layout, cycling params, etc., can be seen in the qPCR Report (see Results).
qPCR - C.gigas COX1 on V.vulnificus exposure cDNA (from 20110311)
Ran a qPCR on all cDNA samples from the V.vulnificus exposure experiment from 20110111. Primers used were Cg_COX1/2_qPCR_F (SR ID: 1192) & Cg_COX1_qPCR_R (SR ID: 1191). Samples were run in duplicate. Master mix calcs are here. Plate layout, cycling params, etc., can be seen in the qPCR Report (see Results).
qPCR - Hard Clam NGS Primer Checks
Ran a qPCR to evaluate a large batch of primers (40 sets) that were ordered per Steven, based off of the most recent SOLiD run (samples submitted 3/10/2011; see Dave’s notebook for more info). Pooled cDNA (2uL from each individual; from 20110511) was used. Master mix calcs are here. Plate layout, cycling params, etc. can be found in the qPCR Report (see Results). The list of primers tested is available in the Primer Database and consist of SR IDs 1233 - 1312. For brevity, samples were only labelled with the corresponding contig number.
qPCR - Lexie’s QPX Temp & Tissue Experiment (see Lexies Notebook 4/26/2011)
Ran qPCR with Lexie’s cDNA samples from this experiment with the following primer sets in order to better evaluate her biological reps:
qPCR - Emma’s New 3KDSqPCR Primers
Due to previous contamination issues with Emma’s primers, Emma asked me to order new primers, reconstitute them and run a qPCR for her to see if we could eliminate her contamination issues with this primer set. cDNA template was supplied by Emma (from 2/2/11) and was from a C.gigas 3hr Vibrio vulnificus challenge. Samples were run in duplicate, as requested. Master mix calcs are here. Plate layout, cycling params, etc. can be found in the qPCR Report (see Results). Primer set used was:
Reverse Transcription - Hard Clam Gill DNased RNA (from 20110509)
Performed reverse transcription on DNased RNA from the hard clam vibrio tubiashii challenge experiment (see Dave’s Notebook 5/2/2011), following the Promega M-MLV RT protocol with ~1ug of DNAsed RNA. Master mix calcs are here. Reactions were done in a plate. cDNA was diluted 1:4 with H2O.
DNase - Hard Clam Gill RNA (from earlier today)
DNased 5ug of RNA from each sample with Ambion’s Turbo DNA-free Kit, according to the manufacturer’s rigorous protocol. Samples were spec’d and stored @ -80C in the “Hard Clam V.t. Experiment RNA” box.
RNA Isolation - Hard Clam Gill Tissue from Vibrio Experiment (see Dave’s Notebook 5/2/2011)
Isolated RNA in 1mL of Tri-Reagent according to the manufacturer’s protocol. Also, finished RNA isolation of samples that were started 20110506. Samples were resuspended in 50uL 0.1%DEPC-H2O and spec’d.
Primer Design - Hard Clam NGS Primers
Designed primers for 40 PCR targets derived from the most recent SOLiD data by Steven(Evernote link) using BatchPrimer3. BatchPrimer3 results are here.
RNA Isolation - Hard Clam Gill Tissue from Vibrio Experiment (see Dave’s Notebook 5/2/2011)
Isolated RNA in 1mL of Tri-Reagent according to manufacturer’s protocol. Samples were precipitated with isopropanol and stored over the weekend @ -20C. Will conclude isolation on Monday. The samples isolated were:
Received - Live Hard Clams From Scott Lindell
Received two bags containing ~24 live clams (didn’t count) in each bag. One bag labeled as “Mashpee Control” and the other “BX Selected.” Clams were stored @ 4C by Steven.
Bacterial Cultures - Colonies Selected from Yesterday’s Colony PCRs
Inoculated 5mL of 1x LB + Kan50 (made by Steven on 3/23/11). Incubated O/N at 37C, 250RPM. Will perform mini preps tomorrow. The following samples were selected:
Mini Preps - Liquid Cultures from yesterday
Mini prepped 3mLs of each culture, according to Qiagen protocol. Samples were eluted with 50uL of Buffer EB.
Bacterial Cultures - Colonies Selected by Steven from Steven’s Re-Streaked Plate
Inoculated 5mL of 1x LB + Kan50 (made by Steven on 3/23/11). Incubated O/N @ 37C, 250RPM. Will perform mini preps tomorrow. The following samples were selected (red text on the plate):
Colony PCR - Colonies from COX1 Genomic Cloning (from 20110411)
Ran colony PCR on various colonies produced from cloning on 20110411. All colonies were picked, re-streaked on Kan50 plate(s) and PCR’d. Master mix calcs are here. Cycling params:
Colony PCR - 5’RACE Colony: COX2 (repeat of yesterday’s PCR)
Repeated yesterday’s PCR on the re-streaked colony in order to run the product on a gel with a more appropriate ladder. See yesterday’s entry for all PCR info.
Colony PCR - 5’ RACE Colony: COX2
One white colony (marked with arrow in image linked) from the two plates from Steven’s cloning (from yesterday) was picked, restreaked on a new Kan50 plate (no X-gal) and PCR’d.
Colony PCR - 5’ RACE Colonies
Two light blue colonies (there were no white colonies) were picked, restreaked on a new Kan50 plate (no X-gal) and PCR’d.
Ligations - COX1/COX2 PCR Products
Performed ligations/cloning on a variety of COX1 genomic and COX 5’ RACE products using the TOPO TA Cloning Kit (Invitrogen). Used 2uL of gel-purified PCR product in each cloning rxn, 2.5uL of H2O, 1uL of salt solution, and 0.5uL of the Invitrogen pCR2.1 vector. Incubated samples for 5mins at RT. Used 2uL of the cloning reaction to transform TOP10 chemically competent cells (Invitrogen), mixed very gently, incubated on ice for 5mins, heat shocked at 42C for 30s and immediately placed on ice. Added 250uL of SOC Medium and incubated tubes at 37C, 200RPM for 1hr. Plated 50uL of each transformation on LB+Kan plates (made by Steven on unknown date with unknown Kan concentration) containing 40uL of 40mg/mL X-gal. Incubated O/N at 37C. Remaining volume of transformed bacteria were stored @ 4C.
5’/3’ RACE PCRs - Nested PCRs for COX2 Sequence
Due to the failure of the primary PCR on both 5’ and 3’ RACE cDNA libraries (from 20080619) yesterday, will perform nested PCR using a nested GSP designed by Steven (CgPGSRACEsrNGSP1; SR ID:1209). The 5uL of PCR reactions that were set aside yesterday were diluted to 250uL with tricine-EDTA (supplied with the Clontech SMARTer RACE cDNA Amplification Kit) as instructed in the Clontech manual. The master mix and tube layouts were exactly the same as yesterday’s, but instead of using 2.5uL of RACE cDNA library as template, I used 5uL of the diluted PCR reaction. Additionally, Universal Nested Primers were used instead of the Universal Primer Mix (both supplied in the kit). Cycling parameters followed “Program 2” from the Clontech manual for 25 cycles.
5’/3’ RACE PCRs - COX2 Sequence on 5’ & 3’ RACE Libraries (from 20080619)
Ran PCRs on both the 5’ & 3’ RACE libraries created 20080619 with a new COX2 gene-specifc (GSP) primer designed by Steven (CgPGSRACEsrGSP1; SR ID: 1208). Although this primer was designed to obtain additional 5’ sequence, it was used with both 5’ and 3’ libraries as a precaution in case it accidentally designed on the wrong strand. PCR rxn was set up according to the Clontech SMARTer RACE cDNA Amplification Kit. Master mix calcs are here. PCR cycling followed “Program 1” from the Clontech manual for 25 cycles.
qPCR - C.gigas BB/DH cDNA for PROPS (TIMP3(BB) primers)
Performed qPCR using cDNA from 20110311. This was performed for 2 reps with TIMP3(BB) (SR IDs: 1067 & 1106). Master mix calcs are here. Plate layout, cycling params, etc. can be found in the qPCR Report (see Results).
qPCR - C.gigas BB/DH cDNA for PROPS (HMGP primers)
Performed qPCR using cDNA from 20110311. This was performed for 2 reps with HMGP (SR IDs:359 & 360). Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results).
qPCR - C.gigas COX1/COX2 Tissue Distribution
Performed qPCR using pooled cDNA from 20110311. Pooled 2uL from each of the following samples groups: Dg 3hr C, Gill 1hr C, Gill 1hr E, Mantle 3hr C, and Muscle 3hr C. Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results). Primers sets run were:
qPCR - C.gigas BB/DH cDNA for PROPS
Performed qPCR using cDNA from 20110311. This was performed for additional reps for TIMP3(BB) (SR IDs:1067 & 1106) and HMGP (SR IDs:359 & 360). Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results).
Reverse Transcription - C.gigas BB/DH DNased RNA (from 20090507) for PROPS
Performed RT on DNased RNA using Promega MMLV RT and Oligo dT according to manufacturer’s protocol, using 1ug of DNased RNA, but in a 50uL reaction. Due to large number of samples, cDNA was made in PCR plate. Plate layout and calcs are here.
Reverse Transcription - C.gigas DNased RNA (from 20110131) from V.vulnificus Exposure & Tissues (from 20110111)
Performed RT on DNased RNA using Promega MMLV RT and Oligo dT according to manufacturer’s protocol, using 1ug of DNased RNA. Due to large number of samples, cDNA was made in PCR plate. Plate layout and calcs are here. cDNA was diluted 4-fold (to 100uL total volume) based on qPCR done by Emma on 20110202.
SOLiD Sequencing Submission
Submitted the following 8 samples for SOLiD sequencing at HTGU:
mRNA Isolation - Pooled Black Abalone Dg RNA (from Abalone Dg Exp 1)
mRNA was isolated for SOLiD sequencing by HTGU. Made two pools of San Nick RNA (Control and Exposed) using equal amounts (5ug) of each individual sample. Individual samples used can be found here. mRNA was isolated using Ambion’s Micro PolyAPurist Kit according to protocol. Procedure was performed two times on each pool and then EtOH precipitated. Samples were resuspended in 10uL of The RNA Storage Solution provided in the kit and spec’d on the Roberts Lab ND1000. Samples were stored @ -80C in the “Next Gen Sequencing Libraries” box.
3’RACE - C.gigas 3’RACE for COX2
Used Cg_COX2_3’RACE_short (SR ID: 1197) & Cg_COX2_3’RACE_long (SR ID: 1196) and the Clonetech SMART RACE cDNA Amplification Kit (unknown acquisition date) to attempt to acquire more 3’ sequence of the C.gigas COX2 isoform. Used Gigas 3’RACE cDNA (from 20080610).
NanoDrop1000 Comparison - Roberts vs. Young Lab
A previous comparison was performed (see 20110209), but it was determined that the standard DNA being used to test the machines was old/degraded. Lisa ordered a new standard DNA dilutions series (Quant-iT dsDNA Kit; Invitrogen) and these DNAs were used. All DNAs were measured 5 times and were mixed by gently flicking between each measurement. A “blank” was measured between each different [DNA] and, if the reading was > + or - 1ng/uL, the machine was reblanked.
qPCR - Check DNased RNA BB01 for Residual gDNA (from earlier today)
Ran qPCR on DNased RNA from earlier today to verify removal of contaminating gDNA. Used C.gigas 18s primers (SR IDs: 156, 157). 0.5uL (~40ng) of DNased RNA was used for testing. This corresponds, roughly, to the amount of sample that would be carried through to qPCR analysis of cDNA, assuming 1ug of RNA was used to make the cDNA (cDNA = 1000ng RNA/25uL = 40ng/uL, 1uL of cDNA in 25uL qPCR reaction). Positive control sample was ~25ng BB16 gDNA (from 20090519). Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results). RNA was stored @ -80C in “Sam’s -80C Box”.
DNase - C.gigas BB01 (PROPS) RNA (from 20090507)
Since the previous DNase treatment failed for this sample, will repeat but will start with less RNA (5ug instead of 10ug). Need more DNased RNA to finish repeating of PROPS. Some samples had insufficient quantities of DNased RNA remaining in BB01. Used 5ug of RNA and followed Ambion’s “rigorous” protocol, utilizing a total of 2uL of DNAse for each sample. Briefly, samples were incubated @ 37C for 30mins, an additional 1uL of DNase was added to each sample, mixed and incubated for an additional 30mins @ 37C. After finishing protocol, samples were spec’d.
qPCR - Check DNased RNA BB01 for Residual gDNA (from earlier today)
Ran qPCR on DNased RNA from earlier today to verify removal of contaminating gDNA. Used C.gigas 18s primers (SR IDs: 156, 157). 0.75uL (~50ng) of DNased RNA was used for testing. This corresponds, roughly, to the amount of sample that would be carried through to qPCR analysis of cDNA, assuming 1ug of RNA was used to make the cDNA (cDNA = 1000ng RNA/25uL = 40ng/uL, 1uL of cDNA in 25uL qPCR reaction). Positive control sample was ~25ng BB16 gDNA (from 20090519). Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results). RNA was stored @ -80C in “Sam’s -80C Box”.
DNase - C.gigas BB01 from 20110225
Used EtOH precipitated BB01 RNA from 20110225 and followed Ambion’s “rigorous” protocol, utilizing a total of 2uL of DNAse. Briefly, samples were incubated @ 37C for 30mins, an additional 1uL of DNase was added to each sample, mixed and incubated for an additional 30mins @ 37C. After finishing protocol, samples were spec’d.
Ethanol Precpitation - DNased RNA BB01 (from earlier today)
Due to residual gDNA contamination, will EtOH precipitate in order to treat with DNase again. Add 0.5 vols 3M NaAOc (pH=
qPCR - Check DNased RNA BB01 for Residual gDNA (from earlier today)
Ran qPCR on DNased RNA from earlier today to verify removal of contaminating gDNA. Used C.gigas 18s primers (SR IDs: 156, 157). 0.5uL (50ng) of DNased RNA was used for testing. This corresponds, roughly, to the amount of sample that would be carried through to qPCR analysis of cDNA, assuming 1ug of RNA was used to make the cDNA (cDNA = 1000ng RNA/25uL = 40ng/uL, 1uL of cDNA in 25uL qPCR reaction). Positive control sample was ~25ng BB16 gDNA (from 20090519). Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results). RNA was stored @ -80C in “Sam’s -80C Box”.
DNase - C.gigas BB01 from 20110216
Used EtOH precipitated BB01 RNA from 20110216 and followed Ambion’s “rigorous” protocol, utilizing a total of 2uL of DNAse. Briefly, samples were incubated @ 37C for 30mins, an additional 1uL of DNase was added to each sample, mixed and incubated for an additional 30mins @ 37C. After finishing protocol, samples were spec’d.
Ethanol Precipitation - DNased RNA BB01 (from earlier today)
Due to residual gDNA contamination, will EtOH precipitate in order to treat with DNase again. Add 0.5 vols 3M NaAOc (pH = 5.2), 2.5 vols 100% EtOH, mixed and incubated @ -20C for 30mins. Pelleted RNA @ 16,000g, 4C 30mins. Washed RNA with 1mL 70% EtOH (2x due to fear of residual salts from DNase Buffer). Pelleted RNA @ 16,000g, 4C, 15mins. Resuspended RNA in 45uL nuclease-free H2O. Sample was stored @ -80C (in “Sam’s RNA Box #1) until it could be DNased again.
qPCR - Check DNased RNA BB01 & 09 (from earlier today)
Ran qPCR on DNased RNA from earlier today to verify that it was free of contaminating gDNA. Used C.gigas 18s primers (SR IDs: 156, 157). 0.5uL (50ng) of DNased RNA was used for testing. This corresponds, roughly, to the amount of sample that would be carried through to qPCR analysis of cDNA, assuming 1ug of RNA was used to make the cDNA (cDNA = 1000ng RNA/25uL = 40ng/uL, 1uL of cDNA in 25uL qPCR reaction). Positive control sample was ~25ng BB16 gDNA (from 20090519). Master mix calcs are here. Plate layout, cycling params, etc can be found in the qPCR Report (see Results). RNA was stored @ -80C in “Sam’s -80C Box”.
DNase - C.gigas BB/DH (PROPS) RNA (from 20090507)
Need more DNased RNA to finish repeating of PROPS. Some samples had insufficient quantities of DNased RNA remaining in BB01 and BB09. Used 10ug of each RNA and followed Ambion’s “rigorous” protocol, utilizing a total of 2uL of DNAse for each sample. Briefly, samples were incubated @ 37C for 30mins, an additional 1uL of DNase was added to each sample, mixed and incubated for an additional 30mins @ 37C. After finishing protocol, samples were spec’d.
Data Analysis - Young Lab ABI 7300 Calibration Checks
All runs (3 runs were conducted) were created using a master mix containing C.gigas gDNA (either 50ng or 100ng), 1X Promega qPCR Master Mix, 0.2uM each of forward/reverse primers (18s; Roberts SR ID: 156, 157). The master mix was mixed well and 10uL were distributed in each well of ABI plates. Plates were sealed with ABI optical adhesive covers.
qPCR - Test Young Lab qPCR Calibration
This is a repeat of the two runs from yesterday, just to see if there is a correlation between the failed plates being the first of the day or not. Master mix calcs and cycling params are here (these calcs are from yesterday, but were used again for today).
NanoDrop1000 Comparison - Roberts vs. Young Lab
Due to an apparent reduction in assay sensitivity for the Hematodidium qPCR assay, we have decided to determine if the spec readings of the plasmid DNA being used for the standard curves are accurate. Used C.gigas gDNA and the lambda DNA Standard (100ng/uL) included in the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen) that was marked as received 9/1/10. Tested both the Roberts Lab and Young Lab using these DNAs. At least 6 separate measurements were taken of each DNA on each machine. Samples were briefly mixed by flicking the tube 4-5 times prior to each measurement.
qPCR - Test Young Lab qPCR Calibration (Repeat)
This was repeated from earlier today due to the failure of the previous run, but had to use new gDNA since I ran out of the stock I had previously used. Master mix calcs and cycling params are here.
PCR - New C. gigas COX Primers for Sequencing of Isoforms
Used new primers for obtaining bands for additional sequencing of both COX isoforms in C. gigas. Master mix calcs are here. Master mix shorthand (MM##) is described below:
qPCR - Test Young Lab qPCR Calibration (Repeat)
This is a repeat of a run from 20110204. Here’re master mix calcs. This was being repeated to evaluate whether or not the relative differences in Ct values observed on 20110204 are consistent or not across the plate. Cycling params were as follows:
qPCR - Test Young Lab qPCR Calibration (Repeat)
This is a repeat of an earlier run from today, but with a different qPCR plate. [Here’re master mix calcs (bottom half of page)(https://eagle.fish.washington.edu/Arabidopsis/Notebook%20Workup%20Files/20110204-01.jpg). Cycling params were as follows:
qPCR - Test Young Lab qPCR Calibration
Recently, the Young Lab’s ABI 7300 qPCR machine was calibrated. Steven asked me to run a plate and see how well the calibration worked. Ran a plate with C.gigas gDNA and Gigas 18s primers (SR ID: 156 and 157) that are known to amplify gDNA. [Master mix calcs are here (top half of page)(https://eagle.fish.washington.edu/Arabidopsis/Notebook%20Workup%20Files/20110204-01.jpg). Cycling params were as follows:
Genomic PCR - Repeat of C.gigas COX genomic PCR from 20110118
This was repeated to generate more PCR product for sequencing purposes. PCR master mix calcs and cycling params are here. Master mixes 04 and 05 (MM04 and MM05) were repeated to gain more PCR product from the faint 550bp & 1500bp bands(MM04) and 5000bp band (MM05).
DNase - DNase C.gigas RNA from 20110120, 20110121 and 20110124
5ug of RNA was DNased using Ambion’s Turbo DNA-free kit, following the rigorous protocol (0.5uL of DNase for 30 mins then additional 0.5uL of DNase for 30mins). Calcs for DNase reactions are here. RNA was stored @ -80C in Shellfish RNA Boxes 4 and 5. Samples will be spec’d on Monday.
RNA Isolation - Various C.gigas Tissue from 20110111
RNA was isolated in 1mL TriReagent, according to protocol. Samples were resuspended in 50uL 0.1% DEPC-H2O and spec’d. RNA was stored @ -80C in “Shellfish RNA Box #4
RNA Isolation - Various C.gigas Tissue from 20110111
RNA was isolated in 1mL TriReagent, according to protocol. Samples were resuspended in 50uL 0.1% DEPC-H2O and spec’d. RNA was stored @ -80C in “Shellfish RNA Box #4”.
RNA Isolation - Various C.gigas Tissue from 20110111
RNA was isolated in 1mL TriReagent, according to protocol. Samples were resuspended in 50uL 0.1% DEPC-H2O and spec’d. RNA was stored @ -80C in “Shellfish RNA Box #4”.
Genomic PCR - C.gigas cyclooxygenase (COX) genomic sequence
Attempt to obtain full genomic sequence for C.gigas COX. PCR set up/cycling params/etc are here. Primer set combinations(master mixes) are as follows:
Bacterial Dilutions - Determination of Colony Forming Units from Gigas Bacterial challenge (from earlier today)
All dilutions were performed with 1x LB+ 1%NaCl. 100uL were plated of all dilutions (see below) on 1xLB+1%NaCL plates. Plates were incubated O/N @ 37C. Colonies will be counted tomorrow to determine CFU for each sample.
Gigas Bacterial Challenge - 1hr & 3hr Challenges with Vibrio vulnificus
400mL O/N culture (1x LB+1% NaCl, 37C, 150RPM, 1L flask) of V.vulnificus (STRAIN??) and V.tubiashii (Strain: RE22) were pelleted (4300RPM, 25C, Sorvall ST-H750 rotor). Supe was removed and pellets were each resuspended in 50mL sea water. 1mL was taken from each to use for dilutions to determine colony forming units (CFU).
2010
SOLiD - Retrieved SOLiD Library Samples from CEG from 20101213
Retrieved the following SOLiD libraries from the CEG and stored in -80C in the “NGS Libraries” Box.
qPCR - COX qPCR Vibrio Exposure Response Check
Used COX primers (SR IDs 1060, 1061) and cDNA from 20080327, which consisted of 7 control gigas gill and 7 vibrio-exposed (24hrs) gigas gill samples, labeled as C# and VE#, respectively. The experiment was a 24hr. exposure live Vibrio vulnificus, parahaemolyticus Cf = 2.055x10^11 (6.85x10^7 Vibrio cells/oyster). Note: Used a free sample of 2x Brilliant III Ultra Fast SYBR Green QPCR Master Mix (Stratagene) for this qPCR. Mixed components and set up cycling params according to the manufacturer’s recommendation for the BioRad CFX96.
qPCR - COX qPCR Primer Test and Tissue Distribution
Used new cyclooxygenase primers (SR IDs 1060, 1061) to see how they performed and to evaluate tissue distribution. Tissue distribution was evaluated using the following cDNAs made on 10/27/10 from Emma:
Restriction Digestions/Ligations - MS-AFLP
Reaction calculations are here. Samples were mixed and incubated @ 37C O/N (started at 6PM). Mac will take care of them tomorrow morning.
Restriction Digestions - HpaII and MspI on Mac’s C.gigas Samples: Round 1
1st of 2 rounds of digests were performed. Calculations are here. Samples were incubated 37C for 3hrs, heat inactivated @ 80C for 30mins and then stored @ -20C.
EtOH Precipitations - HpaII and MspI 2nd Round Digests from 20101124
Samples were EtOH precipitated, according to protocol. Samples were resuspended in 20uL of Qiagen’s EB and spec’d.
Restriction Digestions - HpaII and MspI on Mac’s C.gigas Samples: Round 2
Continued with 2nd round of digestions from yesterday. All samples were resuspended in 25uL of H2O yesterday, so brought volume up to 44uL with H2O, added 5uL of appropriate 10X Buffer (HpaII = NEB Buffer #4, MspI = NEB Buffer #1), added 1uL of enzyme, incubated 37C for 3hrs. Heat-inactivated all samples @ -80C for 30 mins.
Phenol:Chloroform Extractions and EtOH Precipitations - HapII and MspI digests from yesterday
Restriction digests from yesterday were mixed with equal volume (50uL) of phenol:chloroform:IAA (25:24:1) and centrifuged 16,000g for 5mins @ 4C. Aqueous phase was transferred to a clean tube and an equal volume (50uL) of chloroform was added. Samples were mixed and centrifuged 16,000g for 5mins @ 4C. Aqueous phase was transferred to clean tubes and EtOH precipitated, according to protocol. Samples were resuspended in 25uL of H2O and spec’d.
Restriction Digestions - HpaII and MspI on Mac’s C.gigas gDNA Samples: Round 1
Set up restriction digests for subsequent analysis by methylation specific PCR (MSP). This will be the first of two rounds of digestion with the same enzyme on each sample. Samples and master mixes are here. Samples were incubated 3hr. @ 37C. All samples were heat inactivated at 80C for 30mins and then stored @ -20C.
QPX Washes
Washed 4 day old QPX cultures. 3 isolates (S-1, TD-81, ATCC), two flasks (13mL) of each were washed in the following manner:
gDNA Isolation
Isolated gDNA from gray whale skin, human cheek cells (my own!) and two different species of algae (species 1, species 2) using Qiagen’s DNEasy Blood & Tissue Kit according to protocol. Incubated all samples at 55C for 1hr. Eluted DNA with 50uL of Buffer AE. Spec’d samples on NanoDrop 1000.
EtOH Precipitation - Whale gDNA from 20101022
Precipitated whale gDNA in hopes of producing a sample with a higher concentration. Added 0.1 vols of 3M NaOAc (10uL), 2.5 vols of 100% EtOH (275uL), mixed thoroughly and incubated @ -20C for 1hr. DNA was pelleted by spinning sample @ 16,000g, 30mins, 4C. No visible pellet. Supe was removed, sample was washed with 1mL 75% EtOH, and pelleted by spinning @ 16,000g, 15mins, 4C. Supe was removed, sample resuspended in 10uL nuclease-free H2O and spec’d.
gDNA Isolation
Isolated gDNA from crab (unknown species), starfish exposed to RoundUp (unknown species) and gray whale blubber using Qiagen’s DNEasy Kit, according to manufacturer’s protocol. Tissue was incubated at 55C with Proteinase K for 1hr. gDNA was eluted with 100uL of Buffer AE and spec’d.
Received Hard Clam Samples and Live Clams from MBL
Received 82 gill samples in RNA Later in 3 microfuge tube racks from MBL (Scott Lindell). Samples were catalogued, boxed (1 box) and stored at -80C.
Received Hard Clam Samples from Rutgers
30 gill tissue samples in RNA Later from CA, MA, & MAX each.
qPCR - Test Plate for Opticon 2
Ran a full plate for testing well-to-well consistency (or, inconsistency!) of the Opticon 2, since it’s been behaving poorly lately. This will provide us with an idea of whether or not the oddities that we’ve been witnessing have any effect on our actual data.
Opticon Calibration
Distributed 50uL of FAM calibration dye to wells. Ran out of dye!!
qPCR - Hard Clam Primers on cDNA from yesterday
Performed qPCR on Friedman Lab machine targeting immune-related genes in hard clam. Rough plate layout/master mix calcs are here. qPCR report from Friedman Lab machine is here (PDF) and shows cycling params, plate layout and Cts.
Reverse Transcription - DNased Hard Clam RNA from earlier today
Prepared cDNA using 1ug of RNA from each of the 3 pools (CA, MA, MAX) and processed according to Promega’s M-MLV protocol, using oligo dT primers. Calcs and master mix set up are here. Briefly, RNA was combined with oligo dT primers, denatured @ 70C for 5mins, immediately placed on ice for 2mins, mixed with RT master mix, incubated 1hr @ 42C, 3mins @ 95C, and then stored @ -20C.
DNase - DNasing Hard Clam RNA from yesterday
Pooled 2ug of each sample in each group (MAX, CA, MA) for a total of 6ug of RNA (3 total samples), brought volume up to 50uL and DNased using Ambion’s Turbo DNA-free following the rigorous protocol. Calcs can be seen here. Spec’d:
EtOH Precipitation - Hard Clam RNA from earlier today
RNA was mixed with 0.1 vols of 3M NAOAc (pH = 5.2) and 2.5 vols of 100% EtOH, vortexed and incubated @ -20C for 30mins. RNA was pelleted @ 16,000g, 30mins, 4C. Supe was discarded and pellet was washed with 1mL 70% EtOH. RNA was pelleted @ 16,000g, 15mins, 4C. This was step was repeated a second time. Supe was discarded, the RNA was resuspended in 50uL of 0.1% DEPC-H2O, spec’d and stored @ -80C:
RNA Isolation - Hard Clam Tissues Rec’d from Rutgers on 20100820
RNA was isolated from the following samples using TriReagent, according to protocol:
Package - Hard Clam Samples from MBL
Rec’d package from Scott Lindell @ MBL containing 65 screw cap tubes in a white microtube rack. All tubes are tissues in RNA Later (presumably). One sample (MA4-5) was lost during a brief centrifugation to get tissue sample unstuck from top of tube and in to RNA Later solution. The head of the tube snapped off and the entire tube/sample was obliterated in the rotor. Also, it appears as though all the tubes leaked RNA Later solution during transport. Samples were temporarily stored @ 4C and will be catalogued/transferred to -80C.
Package - Hard Clam Samples from Rutgers
Rec’d package of hard clam samples from Emily @ Rutgers on wet ice. Package contained numerous 1.5mL snap cap tubes separated in to groups in zip lock bags. Stored temporarily @ 4C. Will catalog and then store @ -80C.
qPCR - HpaII/MspI Digests from earlier today
Restriction Digests - Various gigas gDNA from earlier today
Digest master mixes are here. Digests were incubated @ 37C for 2hrs. and then heat inactivated @ 80C for 20mins.
gDNA Isolation - Various gigas samples (continued from yesterday)
Pelleted residual tissue 10mins @ 10,000g @ RT. Transferred supe to new tubes. Precipitated DNA with 0.25mL 100% EtOH. Incubated 3mins @ RT. DNA was pelleted 5mins @ 5000g @ RT. Supe was removed, pellets were washed with 1mL 75% EtOH (x2). Supe was fully removed and the DNAs were resuspended in 300uL 8mM NaOH (made 7/9/10 SJW).
gDNA Isolation - Various gigas samples
Placed ~20mg fragments of tissue in 250uL DNAzol. Added 1.35uL of Proteinase K (Fermentas; 18.5mg/mL) to reach a final concentration of 100ug/mL. Incubated RT, O/N, end-over-end rotation. Will complete DNA isolation tomorrow.
MeDIP - SB/WB Fragmented gDNA EtOH precipitation (continued from 20100702)
Finished EtOH precipitation of MeDIP gDNA. Samples were pelleted 16,000g, 4C, 30mins. Supe was discarded. Washed with 1mL 70% EtOH, pelleted 16,000g, 4C, 15mins. Supe discarded. MeDIP DNA was resuspended in 100uL of TE (pH = 8.5). Wash samples, containing unmethylated DNA, were resuspended/combined in a total of 100uL TE (pH = 8.5). Samples were spec’d:
MeDIP - SB/WB Fragmented gDNA (continued from yesterday)
Continued MeDIP process from yesterday. Protein A/G beads were pelleted XXXXXXXXX, supe transferred to clean tube. Beads were washed 3x in the following fashion, each wash saved to retain unmethylated DNA:
Restriction Digests - Various gigas gDNAs of Mac’s
Performed restriction digests. Made dilutions of all DNAs involved of 25ng/uL. Made enough for a total of 9 digests could be performed on each DNA. This allowed using 10uL of each DNA for each rxn, more mileage out of the lowest concentration sample (R37-01), and allowed for the use of master mixes when preparing the digests. All calculations/dilutions/master mixes can be seen here. Each DNA was digested individually with HpaII, MspI and undigested. Incubated the digests 4hrs @ 37C. After digestion, performed an EtOH precipitation. Added 0.1 vols of 3M NaOAc (pH=5.2), then 2.5 vols of 100% EtOH. Mixed by inversion and incubated 30mins @ -20C. Pelleted DNA 16,000g, 30mins @ 4C. Discarded supe. Washed pellets with 1mL 70% EtOH. Pelleted DNA 16,000g, 15mins, 4C. Discarded supe. Resuspended DNA in 10uL PCR H2O and spec’d.
MeDIP - SB/WB Fragmented gDNA (continued from yesterday)
Continued MeDIP process from yesterday. Added 20uL of Protein A/G Plus Agarose (Santa Cruz Biotech) beads to each sample and continued incubation with rotation @ 4C for 2hrs. Pelleted the Protein A/G beads 3300g, 2mins, 4C.
MeDIP - SB/WB Fragmented gDNA (from 20100625)
After confirming proper fragmentation (~460bp average fragment size) via Bioanalyzer earlier today, began the MeDIP process. Brought fragmented DNA samples up to 350uL with TE. Heated samples @ 95C for 10mins, then incubated on ice 5mins. Added 100uL of 5x MeDIP Buffer (50mM Na2HPO4, 700mM NaCl, 0.25% Triton-X 100), 45uL of TE and 5uL (5ug) of anti-methyl cytidine antibody (Diagenode; 5-mC monoclonal antibody cl. b). Incubated O/N, 4C rotating.
Bioanalyzer - Fragmented SB/WB gDNA (from 20100625)
gDNA Sonication - SB/WB gDNA pools (prep for MeDIP) from 20100618
The previous attempt at sonication (see 20100618) failed, likely due to no using the correct equipment (tubes and Covaris adapter). The two gDNA pools, which had previously been unsuccessfully fragmented on 20100618 (SB and WB) were sonicated using a Covaris S2. Used the guidelines of the manufacturer (listed below) for shearing gDNA to a desired target size (500bp):
Samples Received - Hard Clam samples from Rutgers and MBL
Important Note: These were received while I was out of lab. This notebook entry was added 20101021
gDNA Sonication - SB/WB gDNA pools (prep for MeDIP) from earlier today
The two gDNA pools (SB and WB) were sonicated using a Covaris S2. Used the guidelines of the manufacturer (listed below) for shearing gDNA to a desired target size (500bp):
gDNA Precipitation - SB/WB gDNA pools (prep for MeDIP)
8 gDNA samples from SB were pooled and 8 gDNA samples from WB were pooled, using equal amounts of gDNA from each sample (1250ng) for a total of 10ug (see SB/WB Mac’s MeDIP spreadsheet for specific samples/volumes used in pooling). Since samples were stored in pH-adjusted NaOH (see 20100605), they needed to be precipitated in order to have the gDNA suspended in TE for the downstream steps of methylated DNA immunoprecipitation (MeDIP). 10% 3M sodium acetate (pH = 5.2) was added to each tube, then 2.5 vols of 100% EtOH and mixed. Samples were incubated @ -20C for 30mins. DNA was pelleted by spinning 16,000g for 30mins @ 4C. Supe was discarded. Pellets were washed with 1mL 70% EtOH and then pelleted @ 16,000g for 10mins @ 4C. Supe was discarded and gDNA was resuspended in 120uL of TE (pH = 8.0) and spec’d.
gDNA Isolation - Mac gigas larvae samples: control larvae 6.7.10 and 5-aza tr larvae 6.7.10
Continued gDNA isolation of the above mentioned larvae samples that was started by Mac yesterday. Amount of larvae in tubes looked disproportionately large, relative to the amount of DNAzol used in the O/N Proteinase K digestion(~500uL) so I added and additional 500uL of DNAzol to each of the two samples and gently pipetted a few times to mix.
gDNA Isolation - Mac gigas gill samples (continued from yesterday)
Continued with gDNA isolation from yesterday’s samples. Samples were gently pipetted up and down to further dissolve remaining tissue, although tissue did not dissolve entirely. Pelleted residual tissue 10mins @ 10,000g @ RT. Transferred supe to new tubes. Precipitated DNA with 0.25mL 100% EtOH. Incubated 3mins @ RT. DNA was pelleted 5mins @ 5000g @ RT. Supe was removed, pellets were washed with 1mL 75% EtOH (x2). Supe was fully removed and the pellets were resuspended in 200uL 8mM NaOH (made by Amanda Davis 5/20/10).
gDNA Isolation - Mac gigas gill samples
Set up gDNA isolation from the following samples:
gDNA Isolation - Mac gigas gill samples (continued from yesterday)
Continued with gDNA isolation from yesterday’s samples. Additionally, isolated gDNA from R51 01, but homogenized the tissue (using disposable 1.5mL mortar/pestle) in 0.5mL of DNAzol and topped off to 1.0mL. All 3 samples were gently pipetted up and down to further dissolve the tissue. For those samples that were subjected to Proteinase K digestion, I transferred 100uL of the solution to a new tube containing 1mL of DNAzol, [as described in the DNAzol protocol (see “Notes, #5” part of protocol)(https://www.mrcgene.com/dnazol.htm). Tubes were incubated 10mins @ RT.
Package Rec’d - From NOAA in Connecticut
Rec’d 6 15mL conical tubes with liquid cultures of various algae. It appears that we rec’d two of each culture. No note/info included with package. Tubes will be stored @ RT in the styrofoam container they arrived in. Tube labels are listed below:
gDNA Isolation - Mac gigas gill samples
Set up gDNA isolation from the following samples:
SOLiD ePCR/Templated Bead Prep - Lake Trout Lean library
ePCR was performed following ABI’s “full scale” protocol, using 1pM of SOLiD cDNA library.
SOLiD Templated Bead Prep - Yellow perch CT, WB and lake trout Lean libraries (continued from yesterday)
Templated bead preparation was performed according to the “full scale” protocol.
SOLiD ePCRs - Yellow perch CT, WB and lake trout Lean libraries
Performed ePCRs on these samples from DATE, following the “full scale” protocol. A work flow analysis (WFA) run on these samples from the initial ePCRs/templated bead prep (DATE) revealed too many polyclonal beads, thus requiring them to be redone. ePCRs will be performed using 1.0pM (120pg/uL) of the SOLiD cDNA fragment libraries, instead of the 1.5pM (180pg/uL) used previously.
Package - Hard Clam gill tissue/hemolymph in RNA later
Rec’d package from Rutgers (Emily Pearson) containing two large Ziplock bags on “wet” ice, each of those containing smaller bags with sample tubes in them. One large bag contains gill tissue samples and the other large bag contains hemolymph samples. Samples will temporarily be stored @ 4C until they can be catalogued and boxed by Lexie later today.
DNA Isolation - Qiagen Kit Comparison
Note: This information was added 20140407. Yes, you read that correctly.
qPCR - V.tubiashii primers test (Vpt A and Vt IGS)
Utilized to sets of primers obtained from the Friedman Lab: VptA (referred to as “Hasegawa”, even though the reference article calls the primers Vtp A) and Vt IGS (referred to as “Lee” primers, presumably from a published article). For template, used “RE22 DNA” that was given to me by Elene. Tube is dated 9/10/09 and has no indication of concentration. Performed qPCR on a set of 10-fold dilutions. Plate layout/qPCR set up is here, along with dilution series used.
Templated Bead Prep SOLiD Libraries - Yellow perch WB, lake trout Lean and Sisco, and herring G/O HWS09 libraries
All libraries were prepped according to ABI’s “full-scale” bead prep protocol. Initial bead counts were performed using a hemocytometer in a 1:200 dilution:
ePCR SOLiD Libraries - Lake Trout Sisco and Herring G/O HPWS09 libraries (from 20100408)
ePCR was performed for the above three mentioned SOLiD libraries using 1.5pM (180 pg/uL) of cDNA, according to the ABI “full scale” ePCR protocol. ePCRs were stored @ 4C until ready for the emulsion breaking step.
Templated Bead Prep SOLiD Libraries - Abalone CC, CE pools and yellow perch CT, PQ libraries
All libraries were prepped according to ABI’s “full-scale” bead prep protocol. Initial bead counts were performed using a hemocytometer in a 1:200 dilution:
ePCR SOLiD Libraries - Yellow perch PQ, WB and Lake Trout Lean libraries (from 20100408)
ePCR was performed for the above three mentioned SOLiD libraries using 1.5pM (180 pg/uL) of cDNA, according to the ABI “full scale” ePCR protocol. ePCRs were stored @ 4C until ready for the emulsion breaking step.
ePCR SOLiD Libraries - Abalone CC, CE pools and yellow perch CT SOLiD libraries (from 20100408)
Emulsion PCR (ePCR) was performed for the above three mentioned SOLiD libraries using 1.5pM (180 pg/uL) of cDNA, according to the ABI “full scale” ePCR protocol. ePCRs were stored @ 4C until ready for the emulsion breaking step.
cDNA clean up & Bioanalyzer for SOLiD Libraries - Abalone, Yellow Perch, Lake Trout, Herring
Amplified cDNA was cleaned up using the Invitrogen PureLink Micro Kit, but was done so according to Ambion’s Whole Transcriptome Analysis Kit protocol and then spec’d.
Gel Purification & PCR cDNA SOLiD Libraries - Abalone, Yellow Perch, Lake Trout, Herring
cDNA was gel purified according to Ambion’s Whole Transcriptome Analysis Kit. The appropriate regions (100 - 200bp) were excised and cut in to 4, 1x5mm pieces. The two “internal” pieces were transferred to individual PCR tubes. The “outer” pieces were transferred together to a 1.5mL snap cap tube and stored @ -20C.
Reverse Transcription SOLiD Libraries - Abalone, Yellow Perch, Lake Trout, Herring
Samples were speedvac’d to dryness and resuspended in 3uL of nuclease-free H2O. Samples were then reversed transcribed according to Ambion’s Whole Transcriptome Analysis Kit. RT master mix set up is here (top portion of sheet).
Hibridizaton/Ligation SOLiD Libraries - Abalone, Yellow Perch, Lake Trout, Herring
All 8 samples were hybridized/ligated according to Ambion’s Whole Transcriptome Analysis Kit using Adaptor A.
Bioanalyzer Total, mRNA and post-fragmentation SOLiD Libraries - Abalone pools
0.5uL of fragmented mRNA from each library (combined with 0.5uL) was run on Agilent Bioanalyzer 2100 using RNA Pico chips/reagents according to Agilent’s protocol.
RNA Precipitation and Fragmentation for SOLiD Libraries - Pooled abalone mRNA (from yesterday)
mRNA was precipitated according to Ambion’s MicroPolyA Purist Kit protocol. Added 0.1vols of ammonium acetate, 2.5vols of 100% EtOH and incubated 30mins @ -80C. Samples were pelleted, washed with 1mL 70% EtOH, pelleted, resuspended in 8uL of nuclease-free H2O and spec’d:
RNA Precipitation & mRNA Isolation for SOLiD Libraries - Pooled abalone total RNA: Carmel control, Carmel exposed
RNA of 8 samples from each group was pooled equally from each individual. RNA was precipitated according to Ambion’s MicroPolyA Purist Kit. Used 0.1 volumes of 3M NaAOc, pH=5.2, 2.5vols of 100% EtOH and incubated 30min @ -80C. Pelleted RNA 16,000g, 30mins. Washed pellet w/70% EtOH and pelleted RNA 16,000g, 15mins. Pellets were resuspended in 50uL nuclease-free H2O and spec’d:
Bioanalyzer for SOLiD Libraries - Fragmented mRNA from Perch, Lake Trout & Herring RNA samples
1uL of each sample from 20100325 was run on the Agilent 2100 Bioanalyzer on a RNA Pico 6000 chip to evaluate RNA quantity and fragmentation.
SOLiD Library Prep - mRNA (perch, lake trout, herring from 20100318) Fragmentation
Fragmented mRNA according to Ambion’s Whole Transcriptome Sequencing Kit. Cleaned up sample using Ribominus Concentration Module (Invitrogen) according to Ambion’s WTS Analysis Kit. Samples were eluted w/20uL of H2O and stored @ -80C. Will Bioanalyze and speedvac at a later date.
Bioanalyzer for SOLiD libraries - Total and mRNA from Perch, Lake Trout & Herring RNA samples (CONTINUED from yesterday)
Total and mRNA aliquots (~5ng/uL) were run on the Agilent Bioanalyzer Pico RNA chips.
mRNA Precipitation for SOLiD - Perch, Lake Trout, & Herring mRNA (CONTINUED from yesterday)
mRNA was pelleted and washed according to Ambion’s MicroPolyA Purist Kit. Pellets were resuspended in 8uL nuclease-free H2O and spec’d. 0.5uL was taken from each sample, transferred to a fresh tube, diluted to ~5ng/uL and stored @ -80C for eventual Bioanalyzer analysis. mRNA samples were stored @ -80C until we receive the Ribominus Concentration Module Kit from Invitrogen (turns out we didn’t have any!) for cleaning up the RNA after fragmentation.
mRNA Isolation for SOLiD - Perch, Lake Trout, and Herring total RNA
Received pooled lean and siscowet RNA from Rick. Samples will be processed immediately for SOLiD fragment libraries. Two 1.5mL snap cap tubes labelled:
PCR - Test Lexie’s Mercenaria 18s contamination issue
Lexie’s PCR with this primer set and a pool of Mercenria cDNA has yielded contamination in all of her waters. Performed two sets of PCR: one with her existing primer working stocks and the other with a fresh aliquot of primer working stocks. Used my own reagents/water. PCR set up and cycling params are here. PCR ran O/N.
qPCR - Mac’s BB/DH cDNA from 20091223
GSTA and DPGN primer sets used. These are duplicates based on initial differences seen between BB and DH expression. qPCR set up and plate layout here.
SOLiD Bead Titration - Herring fragmented cDNA libraries: 2LHKOD09, 4LHTOG09, 6LHPWS09
Continued with templated bead prep from ePCRs for these libraries. Samples were processed according to the ABI “Templated Bead Preparation Guide” following the “full-scale” protocol.
SOLiD ePCRs - Herring cDNA libraries
Herring fragmented cDNA library: 4LHTOG09
SOLiD ePCR - Herring fragmented cDNA library: 2LHKOD09
Using 1.5pM of starting template, based on success of the 3LHSITK09 bead prep (see 20100108).
qPCRs - Mac’s BB/DH cDNA from 20091223
GNRR2 and CALL primer sets used. These are duplicates based on initial differences seen between BB and DH expression. qPCR set up and plate layout is here.
SOLiD Bead Titration - Herring fragmented cDNA library 3LHSITK09 (CONTINUED from ePCR yesterday)
Completed the remainder of the procedure for template bead titration, according to the ABI “Templated Bead Preparation Guide” following the “full-scale” protocol.
SOLiD ePCR - Herring fragmented cDNA library: 3LHSITK09
Due to low yield of templated beads (12x10^6) from the first run through (see SOLiD Bead Titration below), am repeating using 1.5pM of starting template (as opposed to 0.5pM used yesterday). It should be noted that there is NOT a linear relationship between the amount of starting template and the amount of enriched, templated beads one ends up with in this protocol. So, even though I am increasing the starting template by 3-fold, a 3-fold increase in the amount of enriched, templated beads is NOT expected (hopefully it’ll be more than that!).
SOLiD Bead Titration - Herring fragmented cDNA library 3LHSITK09(CONTINUED from ePCR yesterday)
Completed the remainder of the procedure for template bead titration, according to the ABI “Templated Bead Preparation Guide” following the “full-scale” protocol.
qPCRs - Tim’s Adult Gigas gill cDNA (from 20091009)
Duplicates of earlier qPCRs.
SOLiD ePCR - Herring fragmented cDNA library: 3LHSITK09 (from 20091209)
Processed herring fragmented cDNA library 3LHSITK09 (88.ng/uL) according to the ABI “Templated Bead Preparation Guide” following the “full-scale” protocol. Made a 1:1000 dilution (1uL library, 999uL 1x Low TE) = 88.5pg/uL. Mixed 67.8uL of this diluted sample with 32.2uL 1x Low TE to get a final concentration of 60pg/uL (500pM, according to ABI protocol). Oil phase used was previously prepared by Jesse (Seeb lab) in mid-December 2009. This oil phase is stable for 2 months @ 4C.
qPCRs - Tim’s Adult Gigas gill cDNA (from 20091009)
Duplicates of earlier qPCRs.
2009
qPCR - BB & DH cDNA (from 20091223)
qPCR was set up on these cDNAs using the following primers:
qPCRs - BB & DH cDNA (from 20091223)
qPCR was set up on these cDNAs using the following primers:
qPCR - BB & DH cDNA (from 20091223)
qPCR was set up on these cDNAs using the following primers:
Alaska sockeye salmon sampling (with Seebs): Family #13
Juvenile sockeye salmon were subjected to initial heat stress of 18C. 10 fish were weighed, measured and then snap frozen in LN2. Samples were transferred to a freezer box labelled “AL Sockey Family #13” and stored @ -80C. Here is the spreadsheet with all the pertinent info.
qPCR - BB & DH cDNA (from 20091223) and Emma primer sets for testing
qPCR was set up on these cDNAs using the following primers:
qPCRs - BB & DH cDNA (from 20091223)
qPCR was set up on these cDNAs using the following primers:
qPCRs - BB & DH cDNA (from yesterday)
qPCR was set up on these cDNAs using the following primers:
Sequencing - Mac methylation samples, Sam rhodopsin samples, Lisa samples
Samples were submitted for sequencing. Mac prepped all Roberts Lab samples excluding the Opsin VMC gel slice 2 from 20091217-02. The gel slice was purified with Millipore spin columns. The sample was diluted 1:1 with H2O and submitted for sequencing, one time from each direction using the Sep_Op_Fw2/Rv2 primers. Plate layout can be found here on sheet labeled “20091223”.
qPCR - BB & DH cDNA (from earlier today)
qPCR was set up on these cDNAs using HMGP_5’/3’ and SPI_5’/3’ primers. qPCR set up and plate layout can be found here.
Reverse Transcription - BB & DH DNased RNA (from 20090514)
Made a fresh, double batch (50uL rxn instead of 25uL) of cDNA according to Promega MMLV protocol using oligo dT primers. cDNA was put into a plate for faster qPCR loading. cDNA calcs and plate layout are here. Briefly, RNA and oligo dTs were combined, brought up to 37uL, heated @ 70C for 5mins and immediately placed on ice. RT master mix was made (RT master mix calcs are here), 13uL was distributed to each well. Samples were incubated @ 42C for 1hr and then 95C for 5mins.
PCR - Sepia cDNA
This is an exact repeat of the PCR from yesterday, due to inconsistencies between repeated PCRs from yesterday and earlier today. Here’s yesterday’s workup
PCR - Sepia cDNA
This is an exact repeat of the PCR from yesterday, due to inconsistencies between repeated PCRs from yesterday and earlier today. Here’s yesterday’s workup. Samples were stored @ 4C O/N after completion of PCR. Gell was run on 20091217.
PCR - Sepia cDNA and DNased RNA
Set up PCR on recent Sepia cDNA and DNased RNA samples using both S. officianalis_rhodopsin_F, R primers & Sep_op_F2, R2 primers. After yesterday’s intirguing PCR results, we need to confirm that the DNased RNA is free of contaminating gDNA. Additionally, we want to excise bands from the cDNA samples for sequencing. PCR set up is here. RNA was prepped as though making cDNA; diluted 0.2ug of DNased RNA in a final volume of 25uL. Used 1uL of this for PCRs.
PCR - Sepia cDNA (from yesterday)
Set up PCR on recent Sepia cDNA samples using both S. officianalis_rhodopsin_F, R primers & Sep_op_F2, R2 primers. PCR set up is here. Looking back at my old paper (gasp!) notebook from March 2007, I noticed that each primer set required differing amounts of Mg2+. Rhodopsin primers require 3mM Mg2+ in the PCR rxn and the opsin primers require 2mM Mg2+ in the PCR rxn. Mg2+ was added as required and is shown on the PCR set up link above.
Reverse Transcription - Abalone 07:12 DNased RNA (from 20090623)
Spec - DNased Abalone 07:12 RNA
Reverse Transcription - Sepia DNased RNA
Bioanalyzer - Herring Liver cDNA for SOLiD Libraries
Samples were run on the DNA 1000 chip for cDNA smear analysis.
Emulsion PCR - Herring Liver cDNA for SOLiD Libraries
Emulsion PCR was performed with the two inner gel bands cut out earlier today according to the Ambio WTK protocol. PCR was run for 15 cycles. After the PCR, the samples were cleaned up using the Invitrogen PureLink PCR Micro Kit, according to the Ambion WTK protocol. The cleaned up cDNA (referred to as “libraries” from now on) was spec’d prior to running on the Bioanalyzer.
Reverse Transcription - Herring Liver mRNA for SOLiD Libraries
Ligation reactions from yesterday were subject to reverse transcription according to protocol. Master mix workup info is here. Samples were incubated @ 42C, 30mins and then cleaned up using the Qiagen MiniElute PCR Purification Kit, according to the Ambion WTK protocol.
RNA Adapter Hybridization and Ligation - Herring Liver mRNA for SOLiD Libraries
RNA from yesterday was speedvac’d to dryness and resuspended in 3uL of nuclease-free H2O. Samples were mixed with Adaptor Mix A and hybridized according to Ambion WTK protocol. Samples were then ligated for 16hrs @ 16C, according to Ambion WTK protocol.
RNA Fragmentation - Herring Liver mRNA for SOLiD Libraries
Samples from 20091203. 0.5uL was removed from each and transferred to separate tubes and diluted to < 5ng/uL for subsequent Bioanalyzer analysis using the Pico chip. Samples were fragmented using RNase III according to the Ambion WTK protocol and then cleaned up/concentrated using the Invitrogen RiboMinus Concentration Module according to the Ambion WTK protocol.
RNA Isolation - Sepia samples
Isolated RNA from 7 sepia samples received 20091125. Samples were removed from RNA Later, blotted and homogenized in 500uL of TriReagent. 500uL of additional TriReagent was added to the tubes after homogenization. Procedure was followed normally. The sepia retina RNA was isolated separately from the other samples and was resuspended in 100uL of 0.1% DEPC-H2O. The remaining samples were isolated and resuspended in 20uL of 0.1% DEPC-H2O. Nearly all samples had some sort of purple tint to them, ranging from almost black to extremely faint purple hue. The samples were spec’d and then stored @ -80C in Sam’s RNA Box #1.
Sequencing - Dungan Isolates, Lake Trout HRM and Emma DD cloning
Submitted 1.5 plates for Sanger sequencing. Dungan isolates prepared by me, Lake Trout HRM prepared by Rony and Emma’s differential display cloning samples prepared by her. All primers were prepped by me. See the sequencing log for samples and plate layout.
mRNA Precipitation - Herring Liver mRNA for SOLiD Libraries (continued from yesterday)
Spun samples 16,000g, 30mins, 4C. Discarded supe, quick spun tubes, removed residual supe, washed with 1mL 70% EtOH. Spun samples 16,000g, 15mins, 4C. Discarded supe, quick spun tubes, removed residual supe, resuspended in 8.5uL of nuclease-free H2O. Stored @ -80C until ready to proceed with fragmentation for SOLiD libraries.
Hard Clam Challenge - QPX Strain S-1 (continued from yesterday)
All clams appeared to be alive and well. Most had their siphons out when I arrived to start collecting tissues. Clams were shucked after 24hr challenge. Gill and mantle samples were collected in separate 1.5mL snap cap tubes, stored briefly on ice and transferred to -80C in “Hard Clam QPX Challenge 12/2/2009.” box.
RNA Precipitation - Herring Liver RNA for SOLiD Libraries (continued from yesterday)
RNA Precipitation
RNA Precipitation - Herring Liver RNA for SOLiD Libraries
50uL of RNA from each of the following were precipitated O/N @ -20C:
Hard Clam Challenge - QPX Strain S-1
Challenged 2 FL hard clams and 1 BX hard clam with ~100uL of unwashed, 11 day old cultures. 2 FL hard clams and 1 BX hard clam received ~100uL of QPX media, as controls. Injections were done through the hinge using 20G1 needles and aimed for the pericardial cavity. After injections, clams were left out of water for 1.5hrs, then return to small containers of sea water. They will be incubated for 24hrs.
MBL Shipment - Hard Clam gill tissue in RNA Later
Received samples from Scott Lindell today. Two Ziplock bags taped together labelled “11/16/09 Clams scudders.” The bags contain 2mL screw cap tubes with small tissue samples in RNA later. One group of tubes is labelled with FL-3 # and the other group with BX-4 #. Samples will be stored at 4C to be processed later this month.
MBL Shipment - Sepia tissue samples
Received sepia tissue samples in RNA Later from Kendra Buresch. Here’s the list of tissue we received, according to the Post-It with the samples:
MBL Shipment - Hard Clams
Received hard clams from Scott Lindell today. Two bags. One group (4 live clams, 1 empty shell) labelled as “FL” and another group (9 live clams) labelled as “BX.” Clams were transferred to separate plastic shoebox containers with sea water and sand. They were stored at 15C until ready for experiment.
Herring 454 Data
Data from MoGene was received today on two DVDs and one HDD. Data is two runs of two libraries, due to MoGene concerns that the data of the first run looked bad (too few reads). They performed a second run at no charge and provided us with that data as well.
MBL Shipment - MV oysters/cod
Received a shipment of various MV oysters/cod samples from Scott Lindell at MBL. However, these were NOT shipped on dry ice! Samples were put @ -80C. Will be organized at a later date.
PCR - “Unknown” Dungans/Lyons
This is a repeat of yesterday’s set up with LABY primers, but with an annealing temp of 53C in hopes of improving the number of amplicons generated from additional samples. See yesterday’s PCR run for info on samples.
PCR - “Unkown” Dungans/Lyons
This was done on the numbered tubes using the LABY A/Y primers for eventual sequencing. Turns out many of the tubes have some info (other than just a number) on their sides which might provide more information regarding which isolate they actually are. PCR set up is here. Annealing temp 55C.
Hard Clams - Shipment from Rutgers
Received Hard Clam gill samples on “wet ice” in RNA Later from Rutgers. Samples were collected on 11/4/09 (clams held in refrigerator) and preserved (gill tissue collected) on 11/9/09 according to the paper included with the samples. Samples will be stored @ -80C until we are ready to process.
Sequencing - Lake Trout HRM
This is a second submission of 12 individuals from 8 primer sets. The previous sequencing run was botched because I used the combined primer plate instead of a single (forward or reverse) primer for submission.
Oyster CO2/Mechanical Stress - Water quality
See Rachel’s 441 Notebook from 10/28/2009 through 11/4/2009 for experiment info. 500mL of water was collected from the CO2 and the air tanks. Water was vacuum filtered through Watman paper. The Watman paper was allowed to dry over the weekend.
Sample Submissions to MoGene for 454 Analysis - Herring Liver and Testes mRNA
Submitted 400ng of liver mRNA (SR01) and 400ng of testes (gonad) mRNA (SR02) to MoGene. Samples were packed in dry ice and FedEx.
mRNA Isolation - Herring gonad/ovary RNA (from 20091023)
RNA Precipitation
RNA Precipitation - Herring gonad/ovary RNA (from 20091023)
A subset (3 samples from each group) of samples were pooled (see spreadsheet, green-highlighted samples), each providing ~7.5ug of RNA, yielding 112.17uL. 0.1 vols of 3M NaOAC, pH = 5.2 were added to the tube (11.22uL). 2 vols of EtOH (246.8uL) was added to the tube. Tube was vortexed to mix and incubated @ -20C O/N.
mRNA Isolation - Herring Liver RNA (from 20091021)
Isolated mRNA using Ambion’s MicroPolyA Purist Kit according to protocol. Performed two rounds of isolation to decrease residual rRNA carryover that we frequently see after a single round.
RNA Precipitation - Herring Liver Samples
A subset of samples (4 samples from each group) were pooled (see spreadsheet, green-highlighted samples), each providing ~68ug of RNA. The pooled sample was split into two tubes (435uL/tube). 0.1 vols (43.5uL) of 3M NaOAC (pH=5.2) were added, then 2 vols of 100% EtOH (957uL). Tubes were vortexed and incubated @ -80C for 30mins. RNA was pelleted by spinning 16,000g, 30mins, 4C. Supe was removed, pellets washed with 70% EtOH. Tubes were spun 16,000, 10mins, 4C. Supe removed. Pellets were resuspended in 250uL of RNase-free H2O and stored @-80C until Monday for mRNA isolation.
RNA Isolation - Herring Gonad/Ovary Samples
RNA was isolated according to protocol. Pellets were resuspended in 50uL of 0.1%DEPC-H2O, heated @ 55C for 5 mins, spec’d and stored @ -80C in the “Herring RNA Box #1”.
RNA Isolation - Herring Gonad/Ovary Samples
From the Seeb Lab. Homogenized entire gonad/ovary samples in 5mL of TriReagent with the sonicator. In essence, based on the manufacturer’s recommendation, this means the ratio of tissue:TriReagent was ~2x. Transferred 0.5mL of homogenized gonad/ovary sample to 1.5mL snap cap tubes and added an additional 0.5mL of TriReagent, to adjust the ratio of tissue:TriReagent to ~1x. Samples were then stored @ -80C. These will be further processed tomorrow.
RNA Isolation - Herring Liver Samples
RNA was isolated according to protocol. Pellets were resuspended in 200uL of 0.1%DEPC-H2O, heated @ 55C for 5 mins, spec’d and stored @ -80C in the “Herring RNA Box #1”.
RNA Isolation - Herring Liver Samples (LHPWS09 1-6)
From Seeb Lab. Homogenized entire liver samples in 5mL of TriReagent with the Tissue Tearers. In essence, based on the manufacturer’s recommendation, this means the ratio of tissue:TriReagent was ~2x. Transferred 0.5mL of homogenized liver sample to 1.5mL snap cap tubes and added an additional 0.5mL of TriReagent, to adjust the ratio of tissue: TriReagent to ~1x. Samples were then stored @ -80C. These will be further processed once all remaining liver samples have been homogenized inTriReagent.
qPCRs - Tim’s adult gigas challenge cDNA (from 20091009)
Set up qPCR with Cg_P450 primers and TNFRAF3’/5’ primers. Plate layout/setup is here.
qPCR - Tim’s adult gigas challenge cDNA (from 20091009)
Set up qPCR with Cg_HIF1 (hypoxia induced factor 1) primers and prostaglandin E2 primers. Plate layout/setup is here.
qPCR - Tim’s adult gigas challenge cDNA (from today)
Set up qPCR with EF1 primers and IL17 Internal primers. Plate layout/setup is here. Note: gDNA sample used as a “positive” control will NOT amplify with the EF1 primers.
Reverse Transcription - Tim’s adult gigas challenge DNased RNA (from 20091008)
Performed RT rxns. Setup is here. cDNA was subsequently diluted 1:4 (added 75uL H2O to each well) to properly mirror Tim’s previous qPCRs.
qPCR - Tim’s adults gigas challenge re-DNased RNA (from today)
Performed qPCR using q18s primers on re-DNased RNA (1:100 dilution to match final concentration of template after making cDNA). qPCR set up and plate layout are here.
DNase Treatment - Re-DNase of Tim’s adult gigas challenge RNA (from yesterday)
5 samples from yesterday’s treatment still came up on qPCR, so I will re-DNase those 5 samples. Followed standard protocol in Ambion’s Turbo DNA-free kit.
qPCR - Tim’s adults gigas challenge DNased RNA (from today)
Previous qPCR was done incorrectly (wrong primers), so am repeating with the correct primers. Performed qPCR using q18s primers on DNased RNA (1:100 dilution to match final concentration of template after making cDNA). qPCR set up and plate layout are here.
qPCR - Tim’s adults gigas challenge DNased RNA (from today)
Performed qPCR using q18s primers on DNased RNA (1:100 dilution to match final concentration of template after making cDNA). qPCR set up and plate layout are here.
DNase Treatment - Tim’s adult gigas challenge RNA (from 20090930)
Used 5uL of RNA from each sample, brought samples up to 50uL with H2O and treated according to Ambion’s Turbo DNA-free kit. Rigorous protocol was followed (1.5uL DNase initially + 1.5uL additional DNase after 30mins). Transferred treated samples to a PCR plate to facilitate further manipulation of the samples. Will perform qPCR on these samples to make sure treatment worked.
Bioanalyzer Submission - Trout RBC, Colleen’s gigas GE sample, Mac’s DH/BB PCR for SOLiD WTK
Samples were delivered for analysis on the DNA 1000 chip.
Reverse Transcription/cDNA purification/Emulsion PCR - Ligation rxns of trout fragmented RNA for SOLiD WTK (from yesterday)
The four samples from yesterday were prepared according to the Agilent SOLiD WTK protocol. Briefly:
Adapter Ligation - Rick’s trout fragmented control/poly I:C samples for SOLiD WTK
See the Next Gen Seq Library Database for more info. Processed the 4 samples (one set Ribominus only, one set Ribominus + PolyA enriched) according to the Agilent WTK. Briefly:
qPCR - Tim’s adults gigas challenge DNased RNA (from 20091002)
Performed qPCR using q18s primers on DNased RNA (1:100 dilution to match final concentration of template after making cDNA). qPCR set up and plate layout are here.
DNase Treatment - Tim’s adult gigas challenge RNA (from 20090930)
Used 5uL of RNA from each sample, brought samples up to 50uL with H2O and treated according to Ambion’s Turbo DNA-free kit. Transferred treated samples to a PCR plate to facilitate further manipulation of the samples. Will perform qPCR on these samples to make sure treatment worked.
qPCRs - Check gDNA contamination with EF1 & 18s primers in gigas gill RNA (from yesterday)
Ran qPCR on RNA to evaluate gDNA contamination in the samples. Dilutions of the RNA were made at 1:100, which would be the equivalent amount when making cDNA (1:25) and diluting the cDNA (1:4) prior to using in a qPCR rxn. qPCR set up is here with cycling params. Plate layout is here.
RNA Isolation - Tim’s adult gigas challenge samples
RNA was isolated using 500uL of TriReagent for all samples. Samples were resuspended in 100uL of 0.1%DEPC-H2O and spec’d. Samples stored in Tim’s “NAME OF BOX” box.
Bioanalyzer Submission - Rick’s trout RBC samples (various dates)
Submitted Rick’s trout RBC samples to FHRC for bioanalysis using the PicoChip for use with the SOLiD WTK. Submission sheet is here.
RNA Fragmentation - Rick’s trout RBC samples prepped earlier today
EtOH Precipitaiton - Rick’s trout Ribosomoal-depleted RNA for SOLiD WTK (continued from yesterday)
EtOH Precipitation - Rick’s trout Ribosomoal-depleted RNA for SOLiD WTK (from today)
The “control” and “poly I:C” samples prepared earlier today were EtOH precipitated in preparation for fragmentation.
Ribosomal-depleted RNA - Rick’s trout RBC samples for the SOLiD WTK
Prior to starting the procedure, 0.5uL of total RNA was removed from each sample (control, polyI:C), diluted to ~5ng/uL. 1.5uL of each of these was transferred to a 0.5mL snap cap tube for running on the PicoChip on the Bioanalyzer. These were stored @ -80C in the “Bioanalyzer Samples” box.
RNA Fragmentation - Rick’s trout RBC samples prepped earlier today (see below)
Samples were fragmented according to the Whole Transcriptome Kit protocol. Samples were then cleaned up using Invitrogen’s Modified RiboMinus Concentration Module, according to protocol. Briefly:
mRNA Isolation - Rick’s trout RBC samples previously treated with Ribominus Kit (by Mac)
Was given ~0.5ug of each of these two RNA samples and processed them with Ambion’s microPolyA Purist Kit according to protocol. After elution, the samples were EtOH precipitated @ -80C for 30mins, pelleted 30mins 16,000g for 30mins, 4C. Supe removed, RNA washed with 1mL 70% EtOH and spun 10mins 16,000g, 4C. Supe removed. Resusupended in 8uL of The RNA Storage Solution and spec’d.
mRNA Isolation - Gigas BB and DH samples previously treated with Ribominus Kit (by Mac)
Was given ~0.5ug of each of these two RNA samples and processed them with Ambion’s microPolyA Purist Kit according to protocol. After elution, the samples were EtOH precipitated @ -80C for 30mins, pelleted 30mins 16,000g for 30mins, 4C. Supe removed, RNA washed with 1mL 70% EtOH and spun 10mins 16,000g, 4C. Supe removed. Resusupended in 10uL of The RNA Storage Solution and gave back to Mac.
mRNA Isolation - Gigas BB and DH samples previously treated with Ribominus Kit (by Mac)
Was given 1ug of each of these two RNA samples and processed them with Promega’s PolyA Tract Kit according to protocol. After elution, the samples were EtOH precipitated @ -20C for 30mins, pelleted 30mins 16,000g for 30mins, 4C. Supe removed, RNA washed with 1mL 70% EtOH and spun 15mins 16,000g, 4C. Supe removed. Resusupended in 15uL of 0.1% DEPC-H2O and spec’d.
HRMs - Lake Trout SNPs (HRM_white-05 & HRM_white_06)
HRM_white-05
HRMs - Lake Trout SNPs (HRM_white-03 & HRM_white-04)
HRM_white-03
HRM - Lake Trout SNPs (HRM_white-02)
HRM_white-02
HRM - Lake Trout SNPs (HRM-white-01)
The following primers from primer plate LTP01 will be used for analysis of Rick’s Lake Trout DNA plate (from 4/28/2009): A1, C1, H1, B2. So, that’s 4 primer sets x 96 DNA samples = 384. HRM set up is here. A 1:10 dilution plate of Rick’s Lake Trout DNA1 plate (from 4/28/2009) was made for HRM. This means approximately 20ng of DNA used in each rxn. The robot was used to add 1uL of DNA from the 96-well plate to the appropriate wells of the 384-well HRM rxn plate. Plate was spun to collect the DNA at the bottom of the wells. The wells were visually inspected to ensure that each received the DNA. 1uL of DNA was manually added to those wells that did not receive sample (B23, C23).
qPCR - HRM Lake Trout SNP primer test
Tested out the plate of Lake Trout primers (LTP01 - forward and reverse combined) for SNP detection. qPCR was performed using Roche 2x HRM M.M. qPCR set up is here. Cycling params are as follows:
Primers - Lake Trout Primers for HRM
The two primer plates (LTP01F, LTP01R) were received today. Primers (supplied as 10 nmoles of each) will be reconstituted with 100uL of PCR H2O to make a Cf of 100uM. Forward and reverse primers will be combined in a new plate, in equal volumes, to give a Cf of 50uM. These combined primers will be used to test them on pooled lake trout DNA to identify functional primer pairs for use in HRM.
qPCR - Gigas gDNA test of recalibrated Opticon 2
Master mix containing Gigas gDNA will be used to verify that the recalibration did work. qPCR setup/plate layout is here. I’ve made a master mix using Cg_HSP70_F/R primers designed by Mac. gDNA is BB #12 (0.445ug/uL) from 20090519. The gDNA will be added to the master mix.
qPCR - Recalibration of Opticon 2
DNA Precipitation - C.pugetti DNA for JGI submission (continued from yesterday)
Sample was removed from -20C and spun @ 4C, 16,000 x g for 30mins. Supe removed, pellet washed with 1mL 70% EtOH and spun @ 4C, 16,000 x g for 15mins. Supe removed, tube spun briefly and remainder of EtOH removed. Pellet was resuspended in 100uL of 1x TE and spec’d. Sample will be run on a gel according to JGI instructions.
DNA Precipitation - C.pugetti DNA for JGI submission
qPCR - Additional Calibration test of Opticon 2
qPCR - Carita Primer Test for High Resolution Melt (HRM) Curve Analysis
Ran a qPCR on Rick’s Lake Trout DNA from 4/28/2009 using primers in Carita’s CMA01 Primer Plate (Excel file). DNA was pooled (2uL from each sample), spec’d and diluted to 10ng/uL. qPCR set up is here. Plate layout matches Carita’s primer plate layout. Since we’re just looking for positive/negative samples, I ran this on the Opticon 2 despite the recent “problems” we’ve been having with it. Cycling params used with the 2x HRM M.M. are as follows:
qPCR - Calibration test of Opticon 2
Received new FAM calibration reagent. It comes pre-prepared in a 1x PCR buffer (0.3uM), however there is only enough for a single plate (use 50uL/well). Will run plate in Opticon 2. Run according to the calibration protocol in the Opticon 2 manual (p. 10-4).
qPCR - Calibration test of Opticon 2
Due to results of Opticon testing from 20090722, we have acquired FAM Calibartion Dye from Bio-Rad. Although not listed online or on the product itself, Bio-Rad customer service informed me that the concentration = 1mM. The calibration protocol in the Opticon 2 manual (p. 10-4) says to use 0.3uM (Cf) in 50uL. Will follow this info. Made up 15mLs of dye solution and distributed 50uL into each well of 3 plates. Tested all three plates on two different machines (Opticon 2 and Friedman lab’s).
Bacteria - C.pugetti culture (from 20090713)
1L liquid culture (grown for 10 days) was split into two UV-sterilized bottles. Cells were pelleted in a Sorvall T21 centrifuge using the bucket rotor for 30mins @ 4200RPM, 4C. Supe was removed and bacterial pellets were stored @ -20C.
qPCR - Gigas DNA for Opticon testing
Due to some weird anomolies seen during my previous qPCRs with the H.crach RNA/cDNA samples (positive controls produced good fluorescence when tested in Column 1 of the Opticon, but consistently failed to produce virtually any fluorescence when in Column 6 of the Opticon), I’ve decided to check the Opticon’s fluorescence detection.
RT Rxns - H.crach DNased RNA (from 20090623)
DNased RNA was used for cDNA rxns. [Workup of the amount of RNA used in each rxn (472ng/uL) is here (Google Spreadsheet)(https://spreadsheets.google.com/ccc?key=0AmS_90rPaQMzcHdyU1d0MDVMLWphMFdTOHUwVHFqWnc&hl=en), along with previous cDNA batch numbers. Actual RT master mix set up is here. Samples were given to Lisa for qPCR analysis.
NanoDrop - H.crach DNased RNA (from 20090623)
Samples were first spec’d , since they had not been since their DNase treatement.
qPCR - Abalone cDNA (07:12 set from 3/3/2009 by Lisa) and DNased RNA (from 20090623)
Now that we have a solid positive control, I’ll use the H.crach_h-1fg_intron primers to check the existing cDNA and DNased RNA. qPCR plate layout/set up is here. Anneal temp 50C.
qPCR - Abalone gDNA (H.crach 06:7-1)
This is to test (again!) the H.crach_h-1fg_intron primers and obtain a working positive control. This gDNA is from Lisa/Nate. Acquired from them 20090717. Don’t know date/method of isolation, but this should be good, recent gDNA. qPCR plate layout/set up is here. Anneal temp 50C. Ran serial dilutions of the gDNA: undiluted, 1:10 and 1:100.
qPCR - Abalone cDNA (07:12 set from 3/3/2009 by Lisa) and DNased RNA (from 20090623)
This is nearly a repeat of the qPCR earlier today due to the fact that the positive control never amplified. This is being done to check whether or not there is gDNA contamination in these cDNA and DNased RNA. Will use H.crach_h-1fg_intron primers. In hopes of remedying the positive control issue, I have used three sets of gDNA and used 5uL instead of the usual 1uL for their respective reactions. qPCR plate layout/set up is here. Anneal temp 50C.
qPCR - Abalone cDNA (07:12 set from 3/3/2009 by Lisa) and DNased RNA (from 20090623)
This is being done to check whether or not there is gDNA contamination in these cDNA and DNased RNA. Will use H.crach_h-1fg_intron primers. gDNA 07:12-15 was used as a positive control, based on results from yesterday’s qPCR. qPCR plate layout/set up is here. Anneal temp 50C.
qPCR - Abalone gDNA
Used up the remainder of the one positive control gDNA that worked with all the primers in yesterday’s reaction (H.crach_h-1fg_intron, H.iris_actin_intron, H.crach_16s), so need to find a new set of gDNA to use for future positive controls. qPCR plate layout/set up is here. Anneal temp 50C. Used the following gDNA with :
qPCR - Abalone RNA/DNased RNA & “dirty” and “clean” cDNA
This was done to really test the detection methods we’re using for gDNA contamination in our qPCRs. qPCR plate layout/set up is here. Anneal temp 50C.
PCR - Dungan isolate (MIE-14v) gDNA from 20090708
PCR of MIE-14v just to make sure that we can’t get a product from this sample, despite NanoDrop readings suggesting that there’s no DNA. Used both LABY and Euk primer sets. PCR set up is here. Anneal temp 50C.
Bacteria - C.pugetti large culture
qPCR - Abalone gDNA/cDNA
Due to lack of amplification in gDNA samples from 20090710 and 20090708 with either set of intron primers, will repeat with additional gDNA samples to make sure the primers are the problem and not the gDNA. Used the H.iris_actin_intron_Fw/Rv and the H.crach_h-1fg_intron_Fw/Rv primers. PCR setup/plate layout is here. Anneal temp 50C.
qPCR - DNased Abalone Dg RNA from 20090625
Ran qPCR on gDNA (06:50-10) to test new primers (H.iris_actin_intron_Fw/Rv) designed to bind only to a region in an intron of the H.iris actin gene. Hopefully there’s enough homology between H.iris (primer source) and H.cracherodii (template source) for this to work. PCR setup/plate layout is here. Anneal temp 50C.
DNA Precipitation CONTINUED - Dungan MIE-14v gDNA from yesterday
Sample was pelleted by spinning in a microcentrifuge @ max speed, 4C for 30mins. Supe was removed and sample CAREFULLY washed with 1mL 70% EtOH. Sample was spun in a microcentrifuge @ max speed, 4C for 10mins. Supe was removed, sample brought up in 10uL of TE and spec’d.
PCR - Bay/Sea Scallop DNA
An additional attempt to get the actin primers to work for use in screening samples for bay/sea scallop hybrids. The scallop_actin_fw primer was used in conjunction with the following:
qPCR - DNased Abalone Dg RNA from 20090625
Ran qPCR on DNased Abalone Dg RNA (07:12 Set), gDNA (06:50-10) and clean cDNA (from 20090422) using primers (H.crach_h-1fg_intron_Fw/Rv) designed to bind only to a region in an intron of the H.cracherodii hemocyanin gene. PCR setup/plate layout is here. Anneal temp of 50C was used.
DNA Precipitation - Dungan MIE-14v gDNA from earlier today
Added 1/10 volume of 3M NaOAc (pH = 5.2) to sample (10uL) and then 2 volumes of ice cold 100% EtOH to samples (220uL). Mixed thoroughly and incubated O/N @ -20c.
gDNA Isolation - Dungan isolate MIE-14v
Cells stored in EtOH were pelleted 5000g, 10mins, 25C. A brownish smear was present along the inside of the tube after spinning; not really a pellet per se. Supe was removed and cells were washed twice with 1X PBS. The smear was reduced to a pellet after the first wash in PBS. The second wash resulted in a slightly smaller pellet, but a pellet was present nonetheless before proceeding. Cells were subjected to an enzymatic lysis in 180uL of a TE/Triton X-100/lysozyme mixture as described in the Qiagen DNeasy Kit for Gram-Positive Bacteria (and for cells having substantial cell walls). Cells were incubated in this mixture for 30mins @ 37C. 25uL of proteinase K and 200uL of Buffer AL were then added and the mixture was incubated @ 70C for 30mins. Protocol then followed the “normal” steps for isolation of gDNA. Sample was eluted in 100uL of Buffer AE and then spec’d.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_18s_F/R primers on the following samples that had previously come up negative in both reps using the diluted (1:20) cDNA that Mac had made specifically for the 18s runs:
Spec Reading - C.pugetti gDNA from 20090526
A recent email from JGI indicates that they are satisfied with the quality of DNA (as seen on 20090601), however their estimate of the gDNA concentration (42ng/uL) means that we have ~16ug of DNA. They requested 50ug. Based on the gel, their calculations are reasonable. However, the NanoDrop suggests that are sample is ~1350ng/uL! So, I’ve respec’d the sample and did a few dilutions to see how it looked.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_18s_F/R primers on the following samples that had previously come up negative in both reps using the diluted (1:20) cDNA that Mac had made specifically for the 18s runs:
Bacteria - C. pugetti liquid cultures
Inoculated 3 x 5mL Marine Broth + biphenyl crystals (UV sterilized) cultures from frozen stock in 50mL Falcon tubes. Incubated 200RPM, 28C. Will use these are starter cultures to inoculate larger cultures.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_CIAPIN_F/R primers. This is a second rep. Plate layout/PCR set up is here.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_CatY_F/R primers. This is a second rep. Plate layout/PCR set up is here.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_CatL_F/R primers. This is a second rep. Plate layout/PCR set up is here.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_TLR_“short”_F/R primers. This is a second rep. Plate layout/PCR set up is here.
qPCRs - MV hemocyte cDNA from 20090614
qPCR - BgBL Primers
qPCRs - MV hemocyte cDNA from 20090614
qPCR - CysB Primers
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_18s_F/R primers. Plate layout/PCR set up is here. This is a second rep. Used 1:20 cDNA plate made by Mac as template.
qPCR - Abalone Dg DNased RNA from yesterday and earlier today
Check Abalone RNA for residual gDNA contamination after DNase treatment. Ran qPCR with H.crach_16s_SYB_F/R primers. Plate layout/PCR set up here.
DNase Treatment - Abalone Dg RNA
DNase treated Abalone Dg RNA. Followed Ambion’s Turbo DNA-free Kit rigorous protocol. Here are the calcs/workup for the DNase treatment. This used previously untreated RNA.
qPCRs - MV hemocyte cDNA from 20090614
qPCR - CIAPIN Primers
DNase Treatment - Abalone Dg RNA (07:12 set)
DNase treated the 07:12 set of Abalone Dg RNA. Also, respec’d the RNA prior to proceeding. Followed Ambion’s Turbo DNA-free Kit rigorous protocol. Here are the calcs/workup for the DNase treatment. This used previously untreated RNA.
EtOH Precipitation - DNased Abalone Dg RNA from yesterday AND the 07:12 set (DNased by Lisa 20090306)
Due to the excessive number of times that these samples have been DNased, I’m concerned that the buffer is becoming too concentrated. So, I’m performing an EtOH precip on them to clean them up and then proceed with another round of DNase treatment.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_CatL_F/R primers. Plate layout/PCR set up is here.
qPCR - DNased Abalone Dg RNA from earlier today AND the 07:12 set (DNased by Lisa 20090306)
Set up qPCR using H.crach_16s_SYBR_F/R primers. Plate layout/set up is here.
DNase Treatment - Abalone Dg DNased RNA 20090610
This is the 4th time this RNA has been DNased. Performed using Ambion’s Tubro DNA-free Kit. Followed the “rigorous” treatment protocol.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_TLR _short_F/R primers. Plate layout/PCR set up is here.
qPCR - MV hemocyte cDNA from 20090614
Set up qPCR with Cv_BgBL_F/R primers. Plate layout/PCR set up is here.
qPCR - MV hemocyte cDNA: Test Immomix (SYTO13) vs. Strategene SYBR
Due to craziness seen in melting curves, fluorescence, and empty wells from the previous run, will compare SYTO vs. SYBR with select MV cDNAs. Additionally, acquired some qPCR strip caps to use instead of the ABI film. Used Cv_18s_F/R primers. qPCR set up/plate layout is here.
qPCR - MV hemocyte cDNA from yesterday
qPCR set up/plate layout is here. Used Cv_18s_F/R primers to assess samples’ “useability” for future qPCRs. Used an ABI optically clear adhesive film instead of caps. Ran out of appropriate caps.
Reverse Transcription - MV hemocyte DNased RNA from 20090612
Made cDNA using Promega M-MLV RT and oligo dT primers. RT rxn set up is here. Samples were stored @ 4C until ready to use.
qPCR - DNased MV hemocyte RNA from earlier today AND Turbo kit test
qPCR set up/plate layout is here. Used Cv_18s_F/R primers for the MV hemocyte RNA and Gigas_18s_F/R primers for the Turbo kit test. Anneal 55C.
DNase Treatment - MV hemocyte RNA from yesterday
Samples 3326: B23, A25, A22, B14, A21, A10 B22 came up positive for gDNA still. These were retreated according to Ambion protocol with a brand new Turbo DNA-free DNase kit. Additionally, I tested all three existing kits by “spiking” 19uL of H2O with 1uL (~200ng) of gigas gDNA; one tube for each kit and an untreated sample. Will qPCR to see if gDNA removal was successful.
DNase Treatment - MV hemocyte RNA from earlier today
The entire 20uL of RNA were treated with Ambion’s Turbo DNA-free kit according to protocol and spec’d.
RNA Isolation - Martha’s Vineyard (MV) hemocytes
Samples were pelleted at 100g, 4C for 10mins. The supe was mostly removed, leaving ~50uL of supe above the pellets. RNA was isolated according to the TriReagent protocol from the following samples:
qPCR - Re-DNased abalone Dg RNA from earlier today
This is a repeat of the previous qPCR from earlier today, BUT I think I might have used the wrong primers in the earlier qPCR (see below). Set up qPCR with the correct (I’m 100% sure of this) primers. Plate layout/workup is here.
qPCR - Re-DNased abalone Dg RNA from earlier today
Set up qPCR. Plate layout/workup is here.
DNase Treatment - Abalone Dg DNased RNA from 20090605
This is the 3rd time this will be performed on these samples. Hopefully using a different (i.e. new) kit will work. Not sure what’s going on here. Performed according to protocol.
PCR - C.pugetti gDNA from 20090526
This is a repeat of yesterday’s PCR due to the presence of bands in the water-only samples. Will use reagents and universal 16s bacterial primers (27F & 1492R) provide by the Horner-Devine lab in hopes of: 1) getting this two work and, 2) figuring out the source of the contamination.
PCR - C.pugetti gDNA from 20090513 & 20090526
Previous PCRs from 20090601 & 20090602 both showed contamination in the negative control. Suspect that the primer stocks were contaminated due to the usage of older, non-sterile TE for reconstitution. New stocks were received and reconstituted with filter-sterilized TE. Working stocks were made with filter-sterilized Nanopure H2O. All pipettes, tips, tubes, racks were UV-sterilized in the biological hood. The PCR reaction was set up in the biological hood. PCR set up is here. Used universal 16s bacteria primers (27F, 1492R). Sequences from Sara Kelly. Anneal 60C.
qPCR - Re-DNased abalone Dg RNA from earlier today
Done to verify removal of gDNA from RNA. Used H.crach_16s_syb_f/r primers. PCR workup/plate layout is here.
DNase Treatment - Abalone Dg DNased RNA from 20090605
Had to re-treat these samples due to residual gDNA that was NOT eliminated the first time through. Possible old Ambion kit??
Bacteria - C.pugetti culture (1x 1L)
Inoculated 1x 1L of MB + biphenyl crystals from frozen stock. Incubated 28C, 200RPM. MB was autoclaved with the biphenyl crystals.
qPCR - DNased abalone Dg RNA from earlier today
Done to verify removal of gDNA from RNA. Used H.crach_16s_syb_f/r primers. PCR workup/plate layout is here.
DNase Treatment - Abalone Dg RNA isolated yesterday and from 20090518
RNA from yesterday was treated according to Ambion Turbo DNA-free protocol to remove gDNA contamination. Work up is here.
RNA Isolation - Abalone Dg Project Samples
RNA was isolated from the following samples using the MoBio RNA PowerSoil Kit according to protocol:
DNase Treatment - Abalone Dg RNA isolated yesterday
RNA from yesterday was treated according to Ambion Turbo DNA-free protocol to remove gDNA contamination. Work up is here. Will check for residual gDNA contamination once I’ve finished with another set of RNA isolations from the remainder of Abalone Dg tissue.
PCR - C.pugetti DNA from 20090513 & 20090526
This is a repeat of yesterday’s PCR with a fresh working stock in hopes of eliminating the source of contamination seen in the negative control yesterday.
RNA Isolation - Abalone Dg Project
Isolated RNA using the MoBio RNA PowerSoil Kit according to protocol from the following samples:
PCR - C.pugetti DNA from 20090513 & 20090526
Performed PCR as set up here using universal bacterial 16s primers (sequences provided by Sara Kelly).
DNA Gel - JGI QC check of C. pugetti DNA from 20090526

gDNA Isolation - C. pugetti (from 20090518)
Followed JGI “Bacterial genomic DNA isolation using CTAB” protocol(Word doc) with the following notes/changes.
DNA Methylation Test - Gigas site gDNA (BB & DH) from 20090515
Used BB & DH samples #11-17 for procedure. Followed Epigentek’s protocol. My calcs for dilutions/solutions needed are here. All solutions were made fresh before using, except for Diluted GU1 which was made at the beginning of the procedure and stored on ice in a 50mL conical wrapped in aluminum foil. Used 100ng total (50ng/uL) of each sample gDNA. No standards for a standard curve based on speaking with Mac.
RNA Isolation - Abalone Dg Project samples
Isolated RNA from Abalone Dg samples (see below) using MoBio RNA PowerSoil Kit according to protocol. RNA was stored @ -80C.
C.pugetti - Liquid Cultures
gDNA Isolation - Gigas Dermo Samples
Transferred tissue from previously Chelexed samples:
gDNA Isolation - Mac’s BB and DH site samples
Reverse Transcription - Mac’s gigas DNased RNA from 20090512
Performed RT using Promega M-MLV RT according to M-MLV protocol and used 0.5ug oligo dT per ug of RNA on all BB and DH site samples that were negative for gDNA (see qPCR results 20090512). Calculations and work up are here. Samples were set up in a plate to facilitate sample loading in subsequent qPCRs.
gDNA Isolation - C.pugetti
Isolated according to JGI protocol (Word doc). Used 100mL, 8 day old culture inoculated from a plate on 20090505. Resuspended pellets in 740uL of TE and took an OD600 via the NanoDrop. Diluted the sample appropriately to an OD600 ~ = 1.0 in a final volume of 740uL TE (see the last three measurements for OD600 of final dilution).
qPCR - Mac’s gigas DNased RNA from earlier today
Performed qPCR on the DNased oyster RNA from earlier today with Gigas_18s_F/R primers to verify removal of detectable gDNA in the samples, since the qPCR from 20090508 indicated residual gDNA was still present in some of the DNase treated RNA. Plate layout/set up is here.
DNase Treatment (Rigorous!) - Mac’s gigas RNA/Re-DNased RNA from 20090507 & 20090508, respectively
Followed the rigorous protocol for Ambion’s Turbo DNA-free protocol for the following RNAs:
DNA Isolation - Mac’s gigas samples from 20090505 & 20090506
Isolated gDNA according to Molecular Research Center TriReagent protocol from BB#1-20 and DH#1-20. Resuspended DNA in 600uL of 8mM NaOH. Spec.
qPCR - Re-DNased oyster RNA from today
Performed qPCR on the re-DNased oyster RNA from earlier today with Gigas_18s_F/R primers to verify removal of detectable gDNA in the samples, since the initial qPCR from yesterday indicated residual gDNA was still present in the DNase treated RNA. Plate layout/set up can be found here.
DNase Treatment - Oyster RNA from yesterday
Yesterday’s qPCR indicated that all of the RNA still contained gDNA contamination. So, took 10ug of RNA from BB #1-10 and DH#1-10 (calcs/workup BB and DH) and brought the volumes up to 50uL with 0.1% DEPC-H2O. Processed the samples according to Ambion Tubrbo DNA-free protocol. Will proceed with qPCR immediately.
qPCR - DNased oyster RNA from earlier today
Performed qPCR on the DNased RNA to with Gigas_18s_F/R primers to verify removal of detectable gDNA in the samples. Plate layout/set up can be found here.
DNase Treatment - Oyster RNA from today
All of the RNA (50uL) was DNase treated with Ambion’s Turbo DNA-free kit according to protocol. Samples were spec’d.
RNA Isolation - Mac’s oyster tissues (BB and DH) (CONTINUED from yesterday)
Samples were precipitated according to TriReaget protocol. RNA was resuspended in 50uL of 0.1% DEPC-H2O and then heated @ 55C for 10mins to help dissolve the pellets. Samples will be DNase treated.
RNA Isolation - Mac’s oyster tissues (BB and DH) (CONTINUED from yesterday)
Completed the reaminder of the samples (BB#9-20 and DH#1-20) up to the point of precipitation. Isopropanol was added and stored @ -20C. Organic phase was retained for subsequenct gDNA isolation and stored @ 4C.
Bacteria - C. pugetti liquid cultures
RNA Isolation - Mac’s oyster tissues (BB and DH)
Processed BB#1-8 up to the point of precipitation. Added isopropanol and stored @ -20C. Organic phase was retained for subsequent gDNA isolation and stored @ 4C.
Bacteria - C. pugetti liquid cultures
Inoculated a total of 10, 5mL 1x Marine Broth in 50mL conicals. 5 tubes received 1mL of the original culture started on 20090419. 4 tubes received 1mL of the secondary culture (from 20090421). 1 tube was inoculated with a colony from the plate streaked on 20090424. Incubated all tubes @ 28C, 200RPM. Used a higher temp. to encourage faster/more robust growth.
Bacteria - C. pugetti plate (from 20090424)
An additional yellowing has occurred on the plate. This is in accordance with the Dyksterhouse et al. paper. Colonies still barely visible.
Bacteria - C. pugetti plate (from 20090424)
There appears to be growth on the plate. There is a large, bright yellow/chartreuse region on the plate where the streaking was initiated. Additionally, upon close inspection, it appears as though colonies can be seen.
Bacteria - C. pugetti liquid culture
Results: It has now been 8 days since revival of the ATCC freeze dried culture. The media still looks a bit cloudy, but hasn’t really changed since the second or third day post-revival. Of note, there does seem to be an accumulation of clumps in the tube. These may or may not be clumps of cells. Will consult with Steven when he returns. Might need to pass cells and take some ODs to really assess changes in growth as these cells may not be as robust as E. coli or other common lab cultures.
Bacteria - C. pugetti plate
Streaked C. pugetti onto marine broth plate + a few biphenyl crystals and incubated at RT over the weekend.
Reverse Transcription - cDNA from DNased Abalone RNA from 20090420
[Prepared cDNA using an equal amount of RNA from all samples (442.6ng)(https://spreadsheets.google.com/ccc?key=0AmS_90rPaQMzcHdyU1d0MDVMLWphMFdTOHUwVHFqWnc&hl=en). This amount was based on using the maximum allowable volume of RNA for the RT rxn AND the sample with the lowest [RNA] (08:3-20; 24.59ng/uL). cDNA was prepared according to the Promega MMLV RT recommendations. Here is the work up for the cDNA rxns. cDNA was stored @ -20C.
qPCR - Abalone DNased RNA from yesterday
Performed qPCR to evaluate gDNA removal w/ 2x Immomx and SYTO 13. qPCR/plate set up is here.
Bacteria - C. pugettii culture CONTINUED (from 20090419)
Transferred 1mL of the culture to a 50mL conical containing 4mL of 1x Marine Broth and a couple crystals of biphenyl. Kept the cap loosened and incubated at 20C with shaking at 200RPM. Kept the existing culture in the incubator as well. No apparent growth.
gDNA Removal - Abalone RNA from 20090402 and 20090331
Transferred 50uL of RNA to fresh tubes and processed them using the Ambion Turbo DNA-free kit according to the manufacturer’s protocol.
Bacteria - C. pugettii culture
Rehydrated ATCC isolate according to directions. Added 5mL of 1x Marine Broth to a glass culture tube. Used 1mL of this to rehydrate ATCC sample and then added back into the culture tube. Added a few crystals of biphenyl, which had been exposed to UV for ~5mins prior. Incubated at 20C, no shaking. This was done at ~11:30AM.
Sequencing - Dungan isolates
Submitted two plates for sequencing. Each sample two times from each direction.
PCR - Two new Dungan isolates
Repeat of yesterday’s PCR, but with AmpliTaq, less gDNA and 50uL rxn volume. PCR set up is here.
qPCR - Rab7_SYBR primers on abalone RNA and DNased RNA
Attempt to find out if gDNA contamination exists iafter Ambion treatment. Previous test (on 20090414) suggests the QT Kit did not eliminate gDNA. PCR set up and plate layout here. Used Immomix and SYTO 13.
PCR - Two new Dungan isolates from earlier today
Set up PCRs on:
gDNA Isolation - Two new Dungan isolates
Isolated gDNA from the following two samples:
PCR - Test QT Kit with No RT Abalone rxns from 20090408
Anneal 55C. [PCR set up is here (bottom half of sheet)(https://eagle.fish.washington.edu/Arabidopsis/Notebook%20Workup%20Files/20090414-01.jpg).
PCR - Bay/Sea scallop gDNAs
Used higher annealing temps to improve primer specificity, compared to yesterday’s results. PCR set up and plate layout is here.
PCR - Bay/Sea scallop gDNA isolated earlier today
Used 3 sets of reverse primers:
gDNA Isolation - Bay/Sea Scallop and hybrid samples
gDNA was isolated using 500uL of 10% Chelex. Samples were heated @ 95C for 1hr w/periodic vortexing. Samples were then spun 16,000g for 5mins @ 4C. Stored @ -20C in Bay x Sea Scallop Project Box.
PCR - Abalone gDNA/RNA/cDNA w/new TOLLIP primer
Performed a new PCR on the three types of samples listed above with new TOLLIP primers. The new TOLLIP primers (H.discus_806_F/Ab_866_Rv) surround a putative intron. Thus, they will be useful for determining the presence of gDNA contamination in RNA/cDNA. Anneal temp 55C. PCR set up is here .
PCR - Old Dungan isolates #1-35 w/EukA/B primers
Steven had me re-PCR the old Dungan isolates with the new EukA/B primers. Anneal temp 50C. [PCR set up here (bottom half of sheet)(https://eagle.fish.washington.edu/Arabidopsis/Notebook%20Workup%20Files/20090410-01.jpg) .
PCR - Bay/Sea scallop hybrids
Because Rony’s results from her PCR did not match her previous results for the positive controls, I ran the PCRs myself on the controls and the hybrids. Anneal temp 50C. PCR set up, samples, etc. are here.
PCR - Dungan isolates from 20090402 with Euk primers
Did PCR with new Euk primers designed by Steven. Should be one step higher taxonomically. PCR set up is here. Aneal temp 50C.
qPCR - Check DNased abalone RNA (by Lisa) for gDNA
qPCR was performed with 16s_sybr primers on the DNased RNA that Lisa did. Annel temp 55C. Sample set up and plate layout is here.
qPCR - Repeat (modified) of yesterday’s abalone cDNA check
qPCR was performed with 16s_sybr primers on a subset of the “No RT” cDNA rxns from yesterday at both 55C and 60C. Sample set up and plate layout is here.
qPCR - Abalone cDNA (QT) from earlier today
qPCR was performed with 16s_sybr primers on a subset of the cDNA rxns and all of the “No RT” rxns from earlier to detect the presence of contaminating gDNA. qPCR was also performed with the Rab7_sybr primers on a subset of the cDNA rxns to check that the assay would work. The qPCR set up sheet/plate layout is here. Annealing temp = 55C.
cDNA - Abalone RNA from 20090331 & 20090402
cDNA was made from the above RNA samples using the Qiagen Quantitect RT Kit. The samples were laid out in a PCR plate. 274.2ng of RNA was used in the rxn for each sample, based on the lowest concentration RNA sample (08:3-20) to equalize all the samples. The Genomic Wipeout step of the kit requires 2uL of Genomic Wipeout enzyme/buffer to be added to 12uL of an RNA sample, so the calculations were done and can be found here. A check mark on the calculation sheet indicates that the water and then the RNA was added to the appropriate wells. Those with two check marks were used for a “No RT” rxn and thus, have duplicate wells (see plate layout).
PCR - New Dungan isolates
Repeat of PCR from 20090403, but using AmpliTaq and 50C annealing temp. PCR set up is here.
PCR - New Dungan isolates
Ran PCR with GoTaq on the new Dungan isolate gDNA from yesterday. PCR set up is here. 53C annealing temp.
qPCR - Abalone RNA, check for gDNA
Abalone RNA isolated from this week was qPCR’s with 16s primers to determine if gDNA contamination was present. PCR/plate layout is here.
gDNA Isolation - New Dungan isolates
gDNA was isolated from the following using the Qiagen DNEasy Kit:
RNA Isolation - Abalone digestive gland samples
Total RNA was isolated from the following abalone digestive gland samples using the RNA Powersoil Kit, according to their protocol:
RNA Isolation - Abalone digestive gland samples
Total RNA was isolated from the following abalone digestive gland samples using the RNA Powersoil Kit, according to their protocol:
RNA Isolation - Abalone digestive gland samples
Total RNA was isolated from the following abalone digestive gland samples using the RNA Powersoil Kit, according to their protocol:
qPCR - Virginica cDNA (see workup sheet below for more info regarding cDNA)
Set up qPCR using the following Virginica primers provided by Mac:
mRNA Sample Submission Hard clam gill #1 mRNA from 20090313
Submitted four samples (4uL of each) for Agilent Bioanlyzer:
mRNA Isolation - hard clam gill #1 continued from yesterday
Precipitation was continued from yesterday. Samples were resuspended in 25uL of The RNA Storage Solution (from PolyAPursit Kit) and spec’d. Samples were stored @ -80C in Sam’s RNA box.
mRNA Isolation - hard clam gill #1 DNased RNA from today
DNased RNA from earlier today was split into four equal parts (175uL = 39.8ug). Three will be used for mRNA isolation and the fourth will remain as total RNA. Three of these were precipitated according to Ambion PolyAPurist Protocol: 1/10 volume 5M ammonium acetate, 1uL glycogen and 2.5 volumes of 100% EtOH. Incubated @ -80C for 30 mins. One sample was processed with the Promega PolyA Tract kit. The remaining two samples were processed according to PolyAPurist Protocol. Of those two, one of the samples was processed a second time to evaluate the effectiveness of running a sample through the PolyAPurist Protocol twice.
RNA Precipitation - Hard clam gill #1 RNA from 20080819
DNAsed RNA using Ambion Turbo DNA-free kit, following the rigorous procedure. Diluted total RNA to 0.2ug/uL (Vf = 720uL). Added 1uL DNase and incubated the tube @ 37C for 30mins. Added an additional 1uL DNase and continued incubated for 30mins. Added 0.2 volumes of DNase Inactivation Reagent (158.4uL) and incubated at RT for 10mins with periodic mixing. Pelleted inactivation reagent according to protocol and transferred supe (DNA-free RNA) to clean tube.
qPCR - Repeat of qPCR from earlier today with fresh primer working stocks
This is an exact repeat of the qPCR from earlier today, but using a fresh working stock of the Vtub_16s_V3 primers. The plate layout/qPCR workup is here.
qPCR - Repeat of 20090227 qPCR with clean water
This is an exact repeat of the qPCR from Friday, but using a fresh aliquot of water for preparation. The plate layout/qPCR workup is here.
qPCR - New 16s primers for V.tubiashii Control vs. Autoclaved gigas samples (see 20090224)
qPCR was performed using SensiMix/SYBR “kit” with DNAsed RNA samples from 20090224. This qPCR used the new V.tub_16s_V3 primers in hopes of getting better amplification; both in signal intensity and elimination of the double peak seen in the melting curves from 20090224. The plate layou/qPCR workup is here.
qPCR - Replicate of V.tubiashii Control vs. Autoclaved gigas samples (see yesterday)
This is a repeat of the qPCR from yesterday, but without the 16s and OmpW primer sets due to double peaks in melting curves yesterday. Plate layout/qPCR workup is here.
qPCR - V.tubiashii Control vs. Autoclaved gigas samples (see below)
qPCR was performed using SensiMix/SYBR “kit” with DNAsed RNA samples and cDNA made earlier today. The plate layout/qPCR workup is here.
Reverse Transcription - V.tubiashii DNAsed RNA (from yesterday)
Set up the MMLV RT rxns with random primers using ~833ng DNAsed RNA (prepared yesterday) according to the Promega MMLV Product Insert. This procedure is slightly different than what is in our lab protocol for RT rxns. Here is the workup for the rxns. cDNA was stored @ -20C in the “Vibrio” box.
RNA Isolation - V.tubiashii samples from autoclaved gigas exposure (from 20081218)
RNA was isolated from the Control and V.tub+gigas samples from the 0, 1, & 24hr time points using 1mL TriReagent. No visible pellets. Used 20uL of 0.1%DEPC-H2O to resuspend RNA. Incubated @ 55C, 5mins. Spec’d.
Epigenetics Experiment - Gigas treatment
Set up two containers with 3L seawater and 4 gigas in each container. The treated sample contained:
Sample Submission - V.tubiashii Mass Spec
Vibrio samples (2-1 and 2-2) from 20090212 were submitted to the mass spec facility.
Trypsin digestion - Vibrio 2D spots CONTINUED (from yesterday)
Sample prep was continued from yesterday. SpeedVac’d final, pooled elutions for 1.5hrs. Stored tubes @ -80C. Will submit samples 2-1 and 2-2 for mass spec analysis.
Trypsin digestion - Vibrio 2D spots from 20081217
Samples were prepared according to Goodlett Lab protocol and incubated O/N on LabQuake.
mRNA - Submission for Agilent Bioanalyzer
Submitted 4uL (108ng/uL) of mRNA from gigas gill (from DATE!; sometime in late August) to the Center for Array Technologies (CART) at UW. Sample was labelled “SR01”.
mRNA - Precipitation continued from yesterday
Samples were pelleted and washed with 70% EtOH according to Ambion PolyA Purist protocol. Pellets were resuspended in 10uL of The RNA Storage Solution (included in the Ambion PolyA Purist Kit).
mRNA Isolation - Hard Clam gill and hemo RNA
mRNA was isolated according to Ambion PolyA Purist protocol. After mixing samples with resin, samples were incubated @ RT for 1hr. Samples were washed per the protocol. However, the hemo sample was not clearing from the spin columns with the protocol-directed 3 min. spins. The column had to be spun up to 15 mins. in order for the column to clear. :(
RNA - Hard clam hemo RNA (from 20090121)
The two RNA samples from yesterday were precipitated and washed according to the Ambion PolyA Purist protocol and resuspended in 50uL of 0.1% DEPC-H2O.
RNA Isolation - Hard clam hemo (from 20090121)
The 8 hemo samples were pooled and the 4 gonad/d.g. samples were pooled. RNA was isolated. The homo sample was resuspended in 100uL of 0.1%DEPC-H2O and the gonad/d.g. sample was resuspended in 50uL 0.1%DEPC-H2O. RNA was precipitated O/N @ -20C according to Ambion PolyA Purist protocol in preparation for mRNA isolation.
Bleeding/Tissue Collection - Hard Clams
Hemos were collected from the remaining 12 clams as before and stored @ -80C. NOTE: 4 hemocyte samples were extremely cloudy. Possibly not hemos? Maybe gonad/d.g.? Gill tissue was collected from 5 clams and stored @ -80C.
RNA - Precipitation of Hard Clam Hemo RNA from 20090116
RNA isolated on 20090116 was precipitated over the weekend @ -20C. Samples were treated according to Ambion PolyA Purist protocol and resuspended in 100uL of 0.1% DEPC-H2O. Samples were stored in the red “hard clam” box @ -80C.
RNA Isolation - Hard clam gill, hemos
Bleeding - Hard Clams
Bled 7 clams from 20090108 and 20090109. Bled clams using a 23g 1.5 needle on a 3mL syringe. Fluid was gathered and ranged from ~0.4-1.0mL. Hemolymph was transferred to individual 1.5mL snap cap tubes and spun @ 100g for 30mins @ 4C. Most of the supe was removed, but left ~100uL in each tube to avoid disturbing any pellet. Samples were stored @ -80C in the red box with previous hard clam hemo samples.
RNA - Precipitation continued from yesterday
Transferred supe to a fresh tube and added 1mL 70% EtOH to remaining pellet. Spun samples max speed @ 4C 30 mins. Removed supe and washed pellets with 1mL 70% EtOH. Spun max speed 10 mins. Removed supe . Resuspended the “supe” sample in 50uL 0.1%DEPC-H2O and the “pellet” sample in 100uL 0.1%DEPC-H2O.
RNA - Reprecipitation of hard clam RNA from yesterday
Because of the relatively large size of the pellets vs. the amount of RNA, I think another round of precipitation would be best to help remove additional residual salt carryover. Will precipitate O/N according to Ambion PolyA Purist protocol. RNA pellets were resuspended in 250uL of 0.1%DEPC-H2O and precipitated O/N @ -20C.
Bleeding - Hard Clams
Bled 8 clams from 20090108 and 20090109, #4, 6, 8, 15, 16, 17, 21, 26. Bled clams using a 23g 1.5 needle on a 3mL syringe. Fluid was gathered and ranged from ~0.4-1.0mL. Hemolymph was transferred to individual 1.5mL snap cap tubes and spun @ 100g for 30mins @ 4C. Most of the supe was removed, but left ~100uL in each tube to avoid disturbing any pellet. Samples were stored @ -80C in the red box with previous hard clam hemo samples.
RNA Isolation - Hard Clam hemolymph from 20090108, 20090109
1mL of TriReagent was used to isolate RNA from 3 combined tubes of hemolymph. This resulted in 10 total RNA preps. Pellets were resuspended in 100uL of 0.1% DEPC-H2O and pooled into a single tube and NanoDropped.
Bleeding - Hard Clams
Bled 24 hard clams using a 23g 1.5 needle on a 3mL syringe. Fluid was gathered and ranged from ~0.4-1.0mL. Hemolymph was transferred to individual 1.5mL snap cap tubes and spun @ 100g for 30mins @ 4C. Most of the supe was removed, but left ~100uL in each tube to avoid disturbing any pellet. Samples were stored @ -80C in the red box wiht previous hard clam hemo samples. Clams were numbered and transferred to a holding tank. Gill and mantle tissue was collected from 10 of the clams. The collected tissue and the rest of the carcasses were stored @ -80C.
Bleeding - Hard Clams
Bled 6 hard clams using a 23g 1.5 needle on a 3mL syringe. Fluid was gathered and ranged from ~0.4-1.0mL. Hemolymph was transferred to individual 1.5mL snap cap tubes and spun @ 100g for 30mins @ 4C. Most of the supe was removed, but left ~100uL in each tube to avoid disturbing any pellet. Samples were stored @ -80C in the red box with previous hard clam hemo samples. Clams were numbered and transferred to a holding tank.
PCR - Dungan Isolates
Samples (in Chelex) were vortexed and heated @ 95C for 30mins with periodic vortexing. Tubes were spun max speed @ 4C for 2 mins to pellet Chelex. Set up PCR using Immomix master mix. Anealing temp. = 56C. PCR set up here.
PCR - Dungan Isolates
All samples , except xCVC-11t, are already in Chelex. For xCvC-11t, pipetted a shunk of cells/tissue from source tube. Volume of liquid (EtOH) was ~75uL. Added this to screw cap tube containing 300uL of 10% Chelex solution. Vortexed and incubated @ 95C for 30mins. Vortexed and incubated other samples at 95C for 5mins. Set up PCR with AmpliTaq. Anneal temp. = 53C. PCR set up here.
SDS/PAGE, Western Blot - Test of HSP70 Ab on heat stressed shellfish for FISH441
Pacific oysters, a mussel, barnacles and a clam (sp. ?) were transferred from the holding tank to a large beaker with sea water which was placed into a 37C water bath. The shellfish were incubated in this water bath for ~3hrs. Tissues were collected from each, transferred to a 50mL conical tube and immediately placed in a dry ice/ethanol bath:
2008
SDS/PAGE, Western Blot - Test of new Western Breeze Kit & HSP70 Ab for FISH441
RNA Gel - V. tubiashii mRNA samples (from 20081224)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
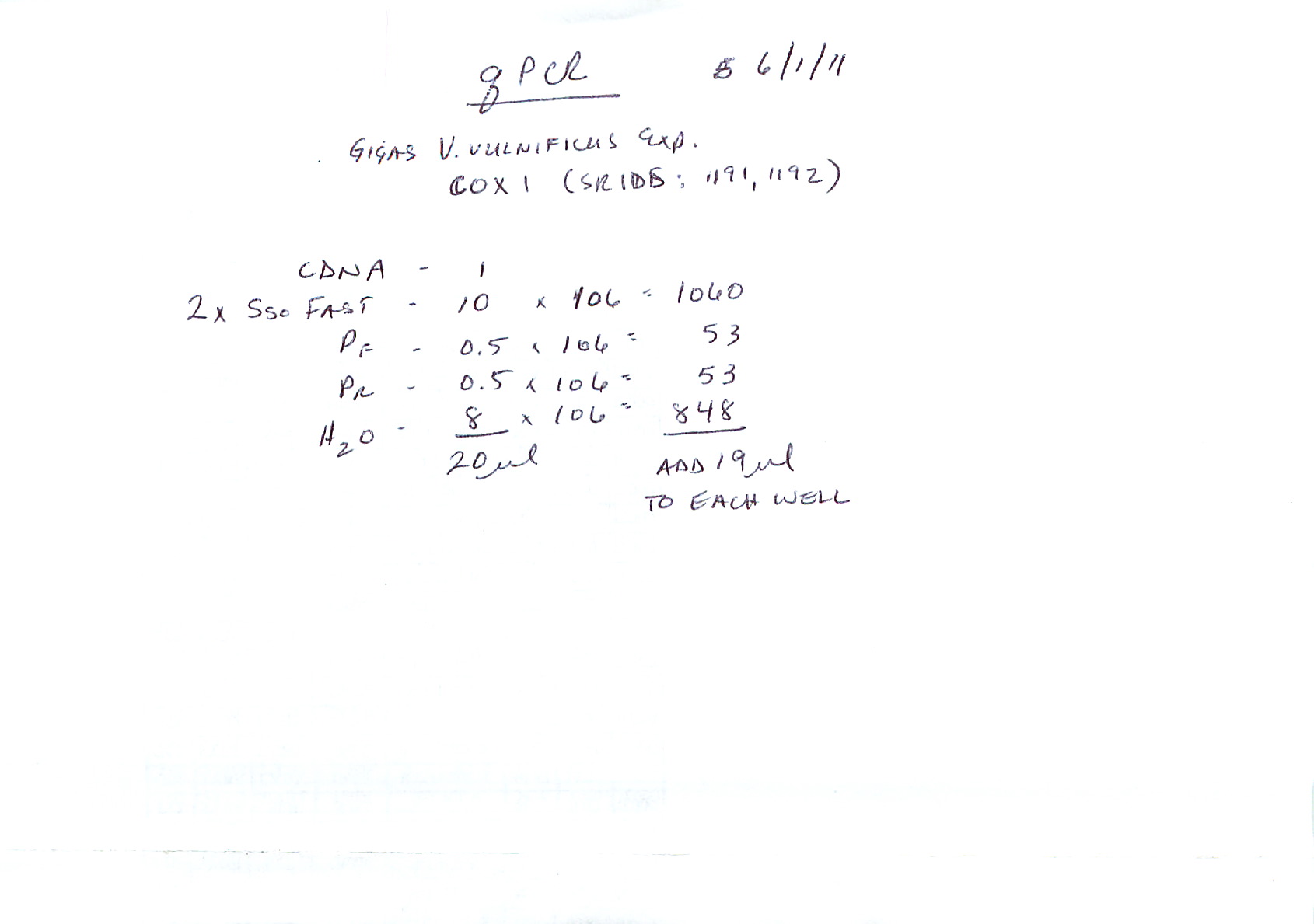{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
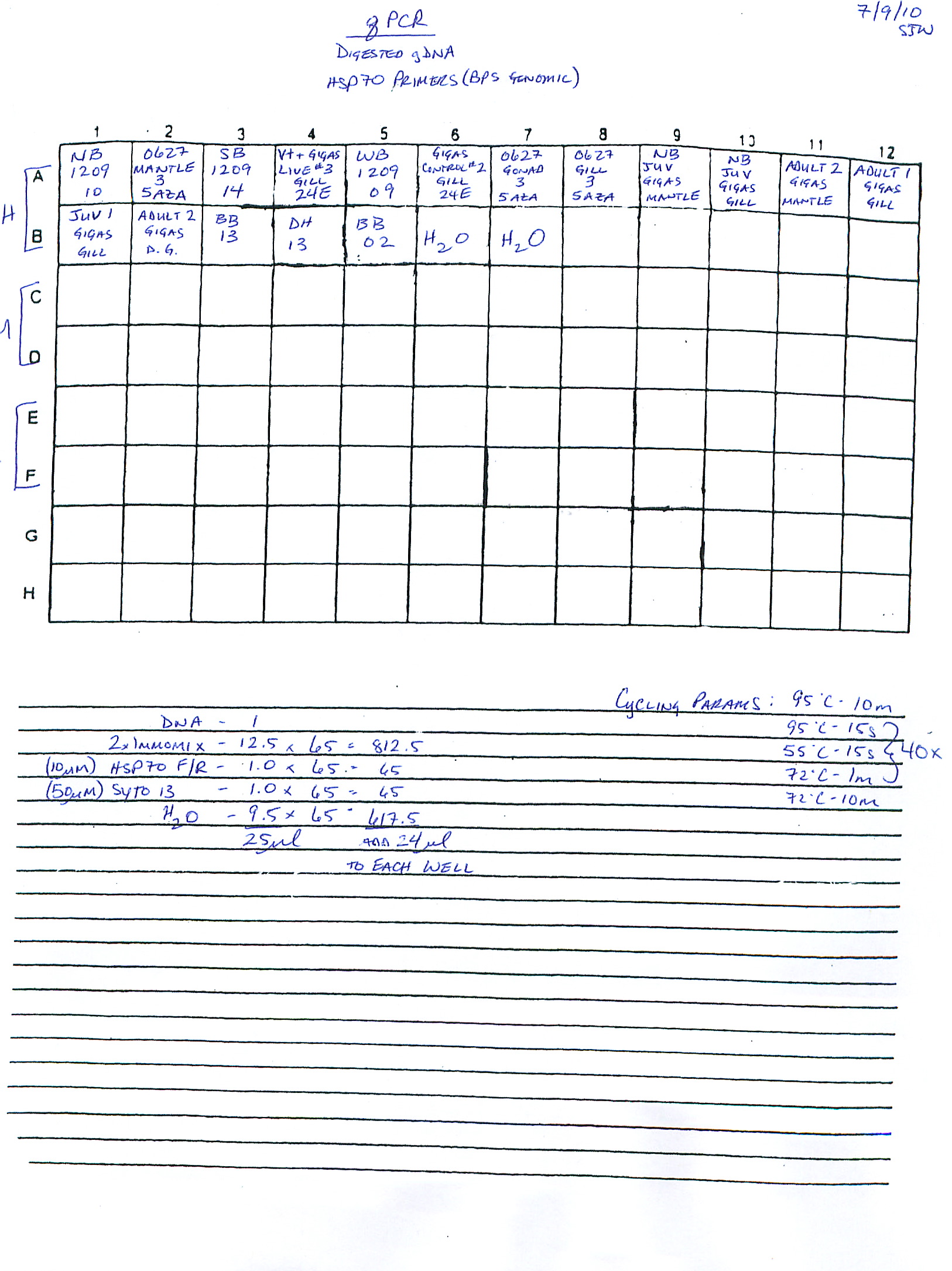{kind=link}
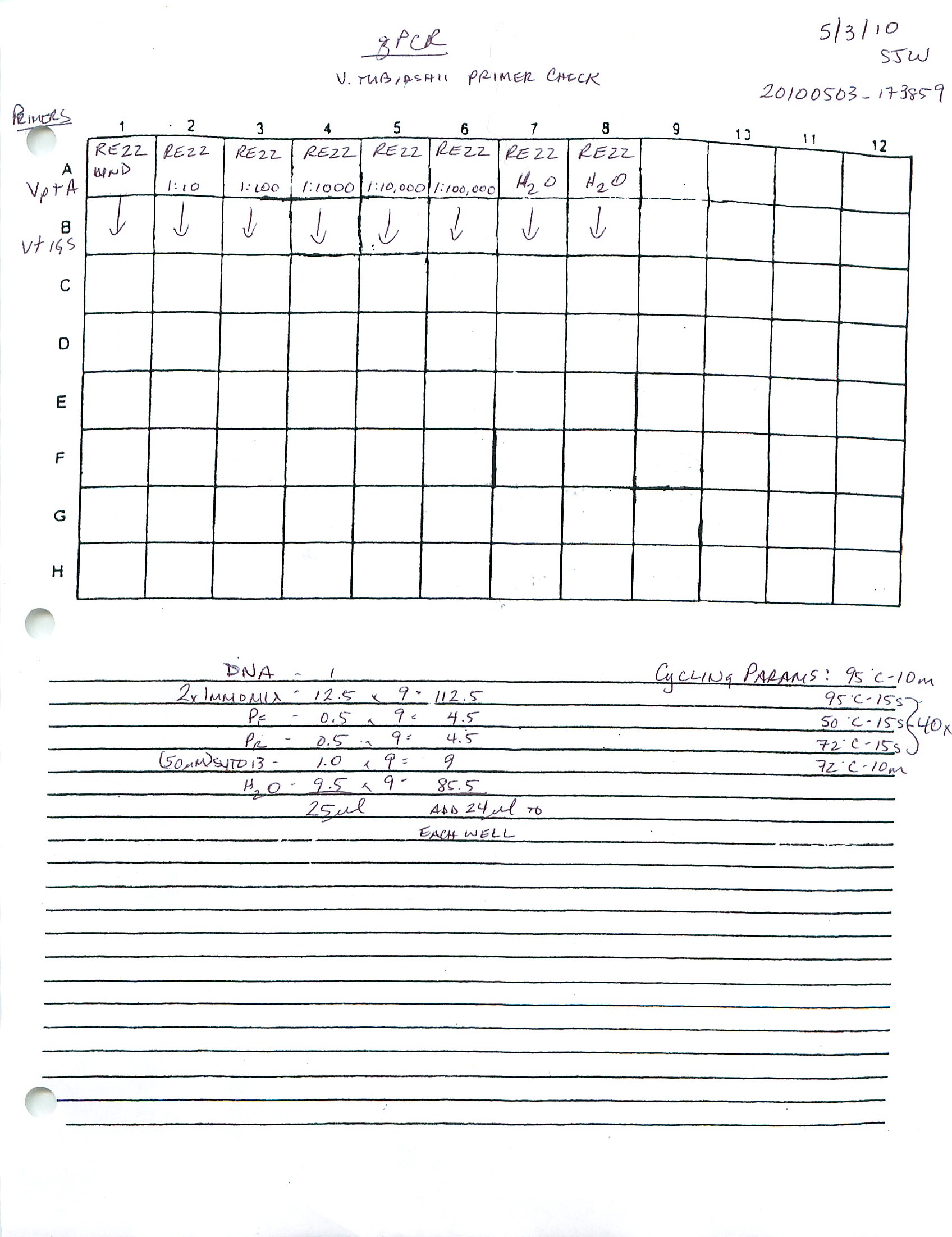{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
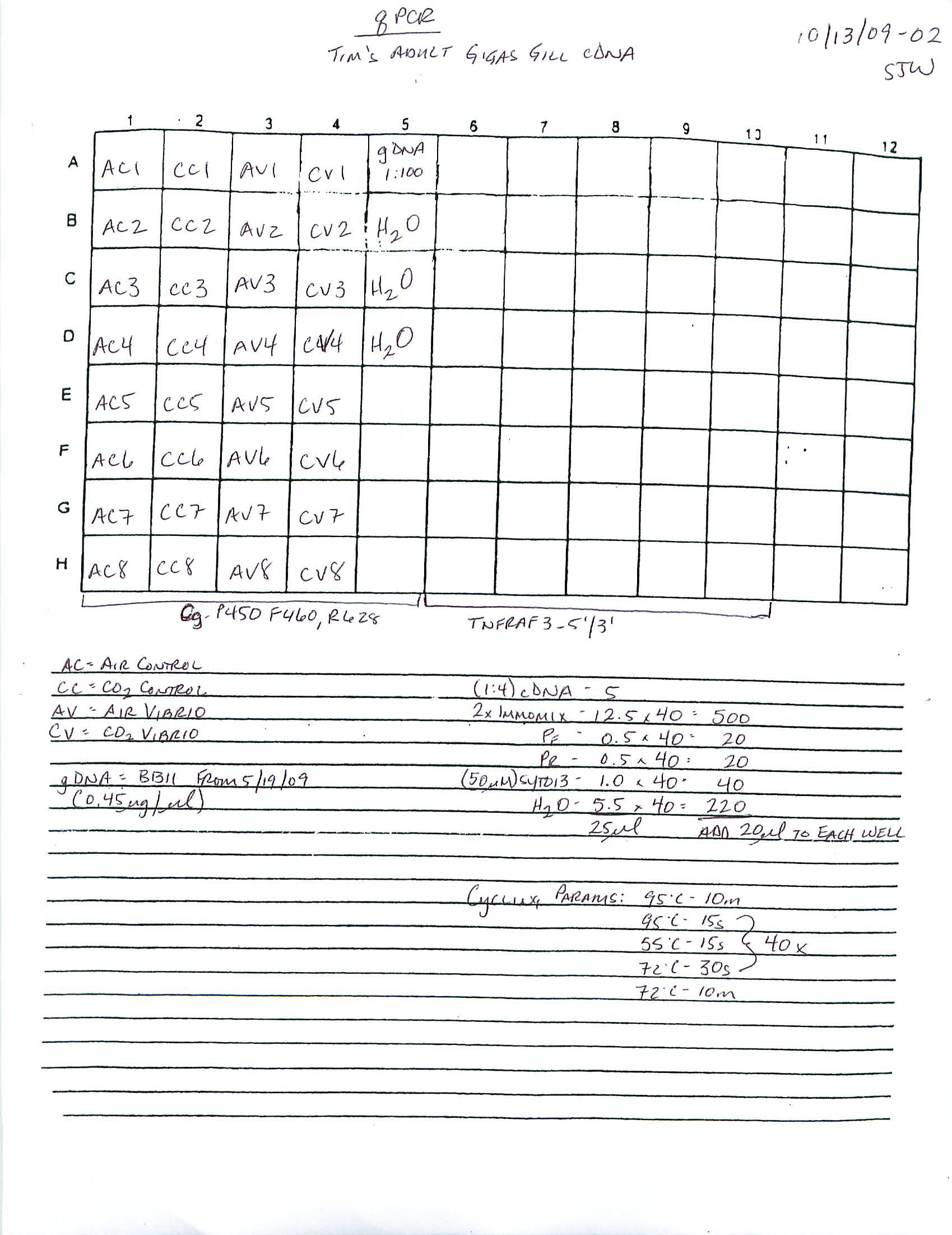{kind=link}
{kind=link}
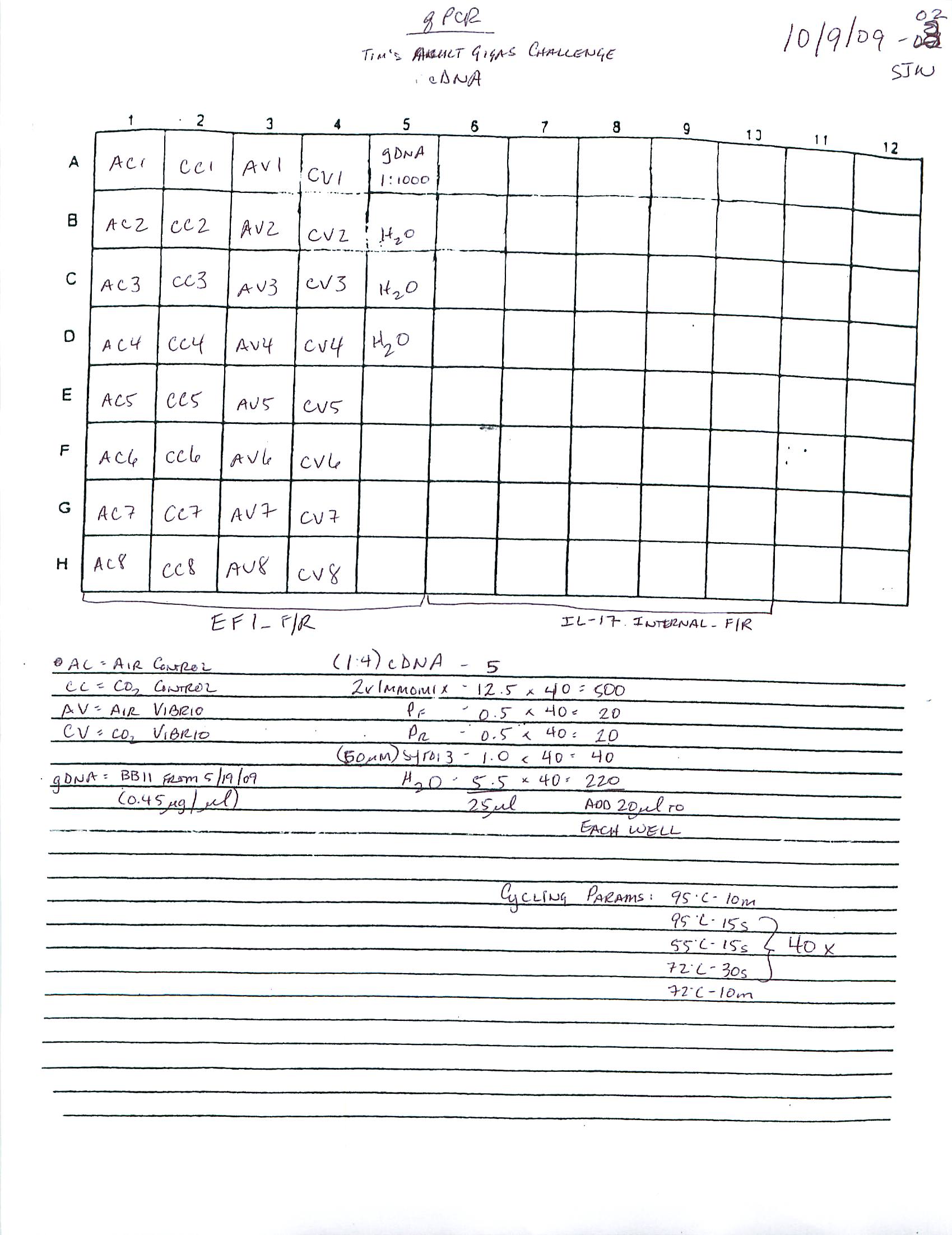{kind=link}
{kind=link}
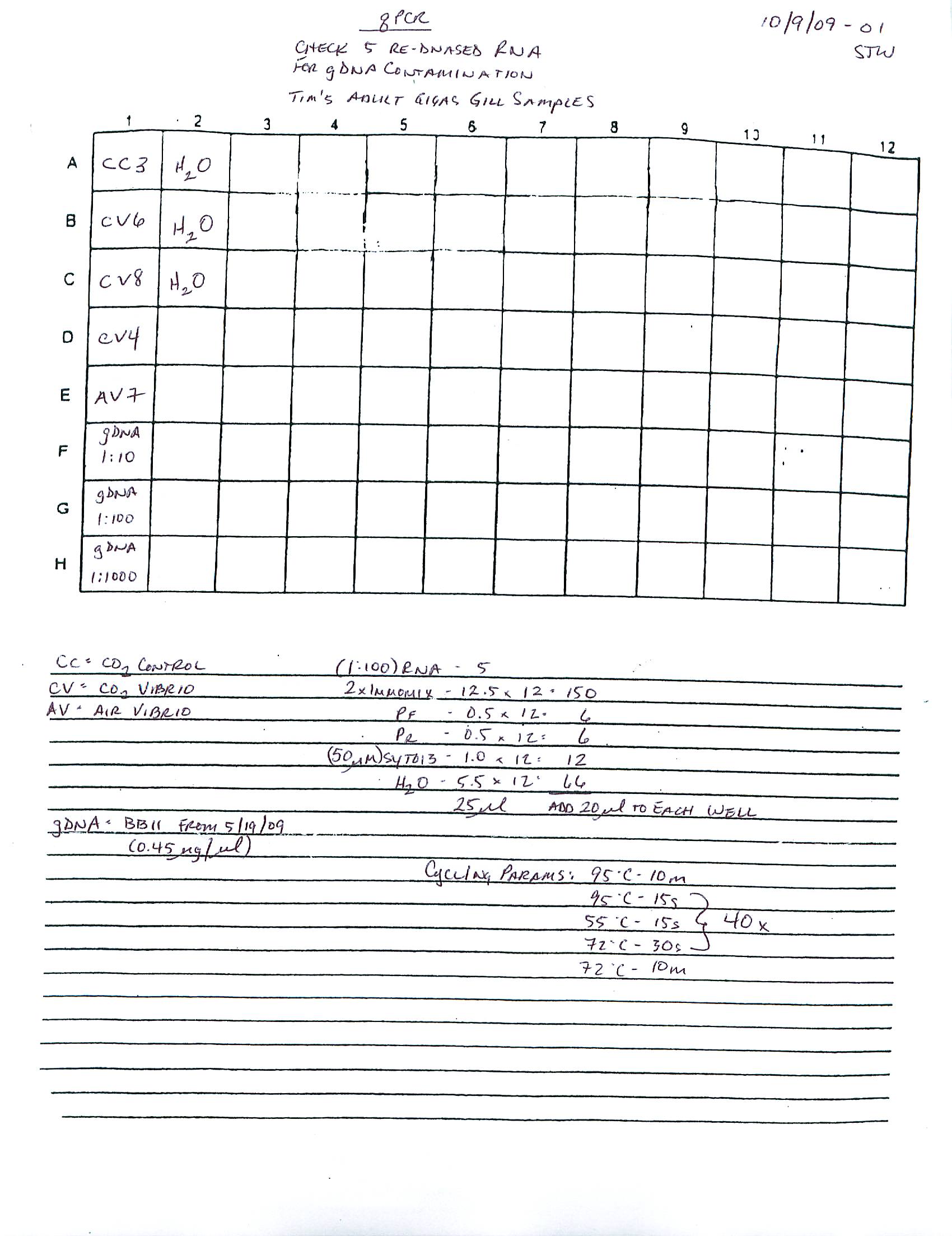{kind=link}
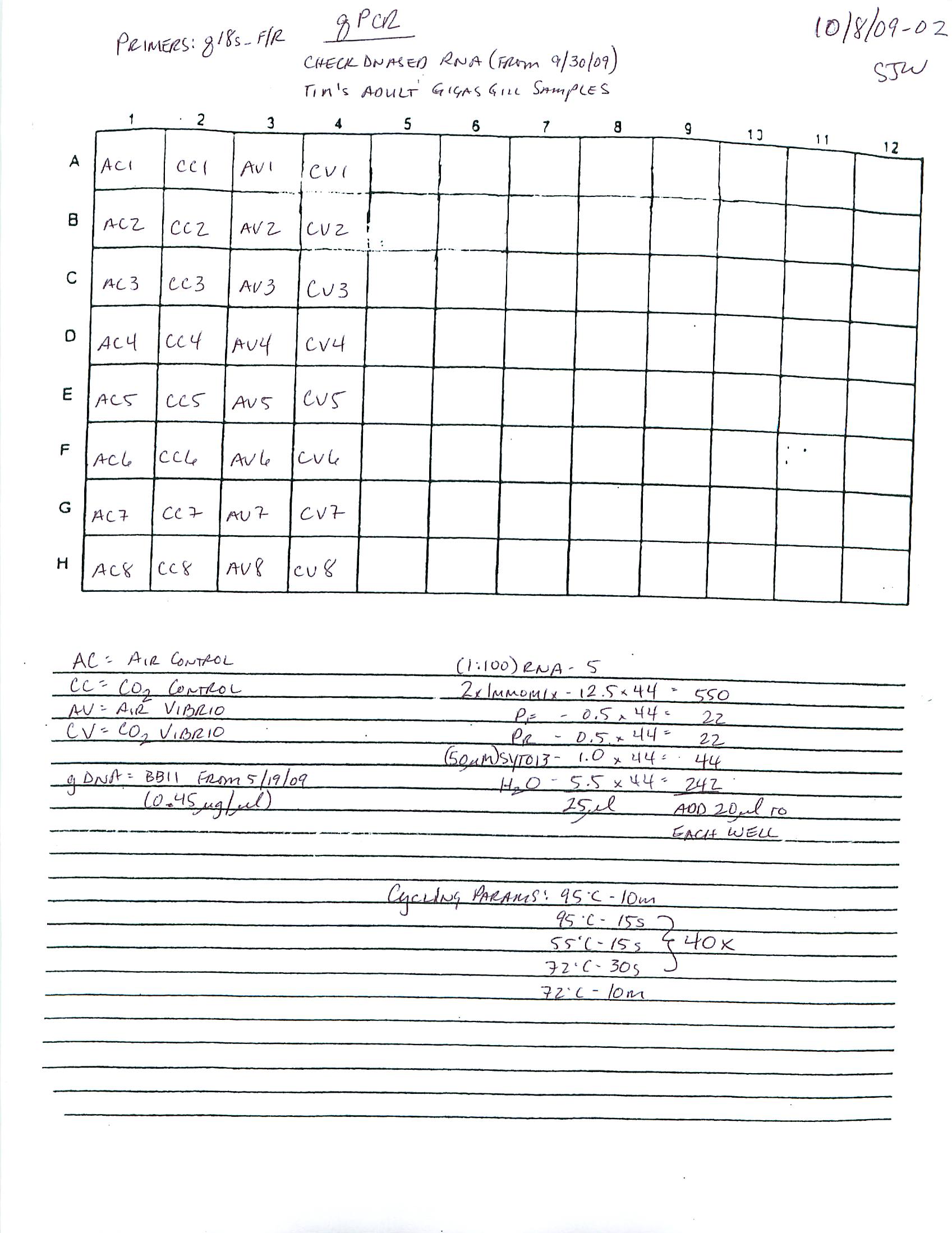{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
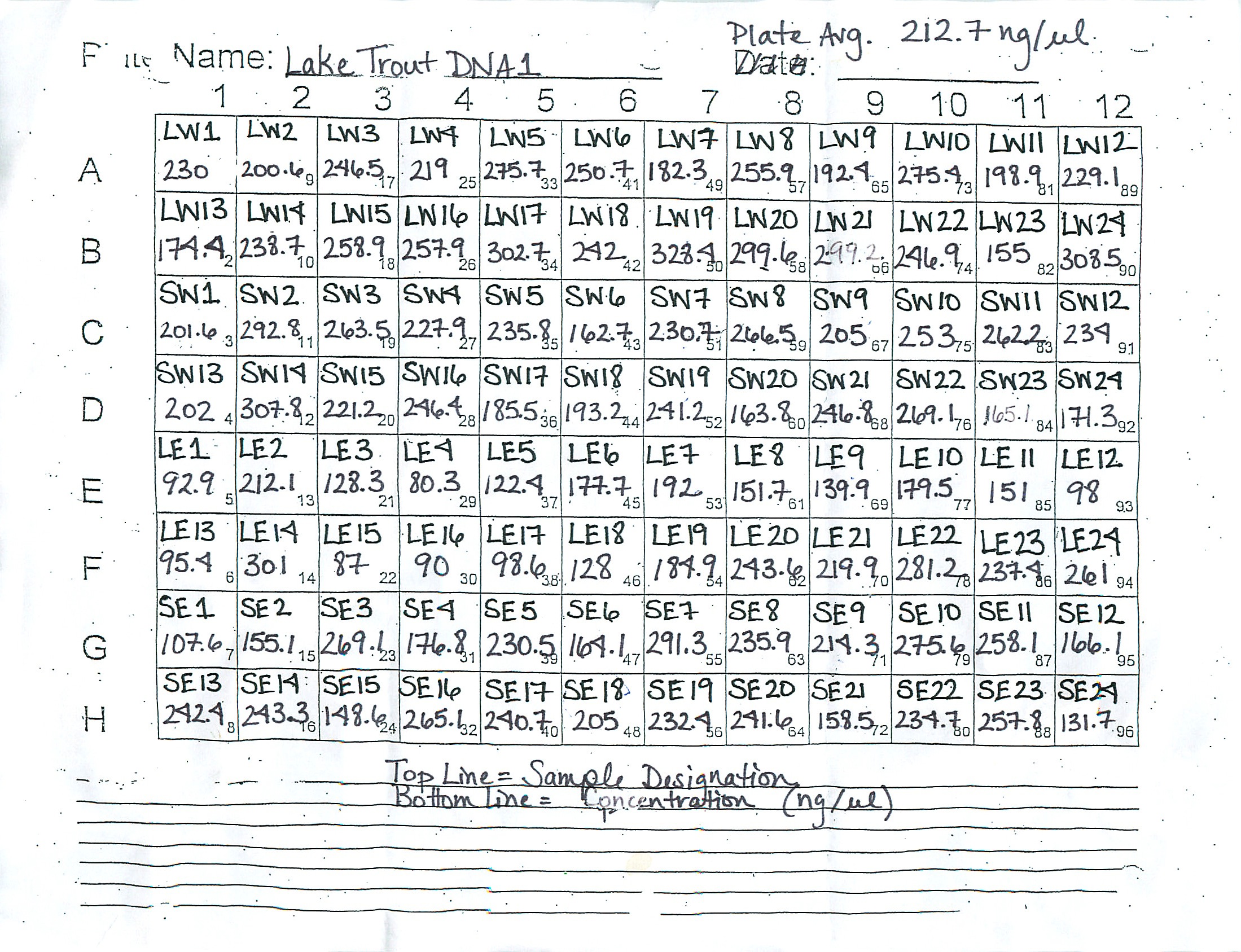{kind=link}
{kind=link}
{kind=link}
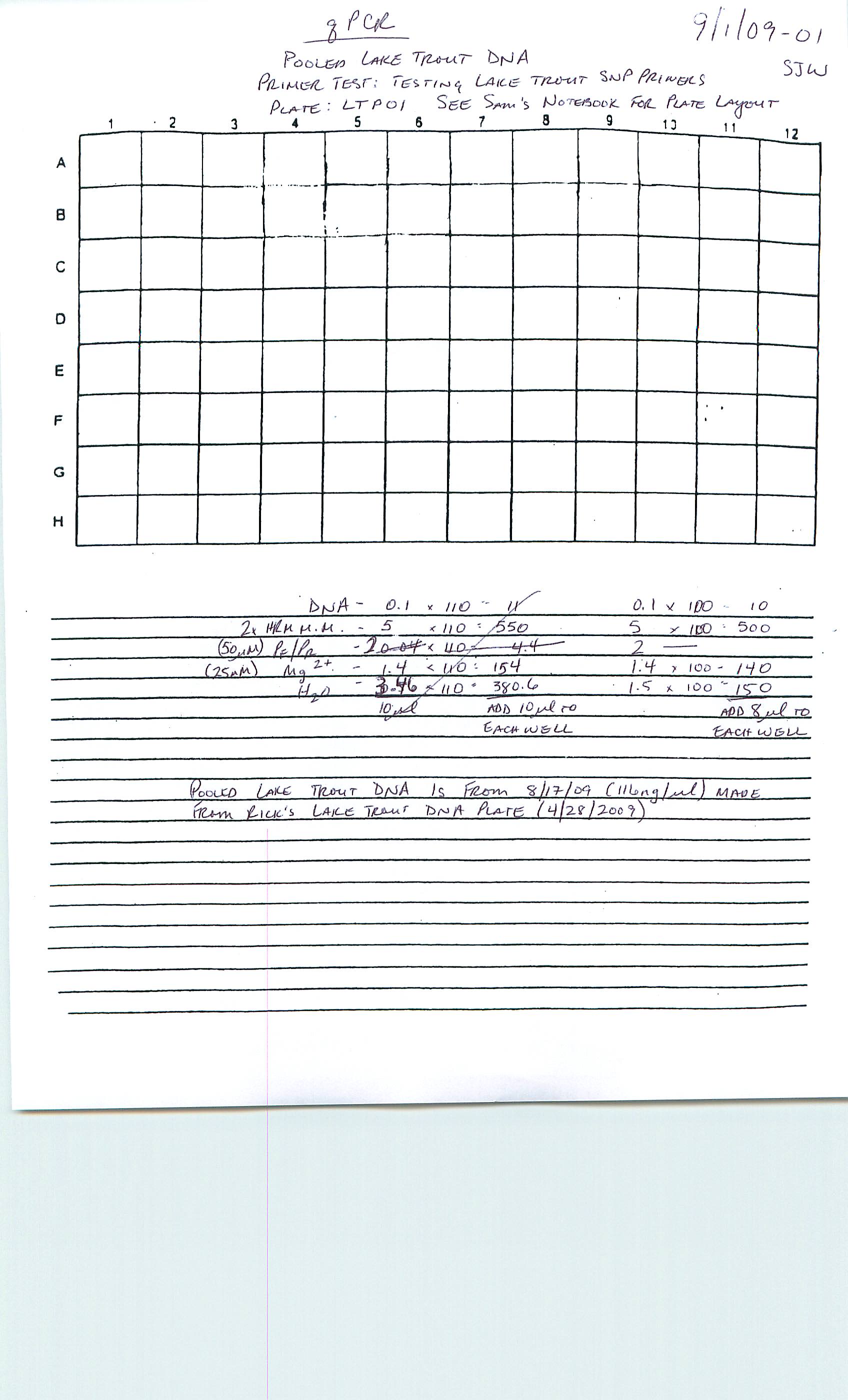{kind=link}
{kind=link}
{kind=link}
{kind=link}
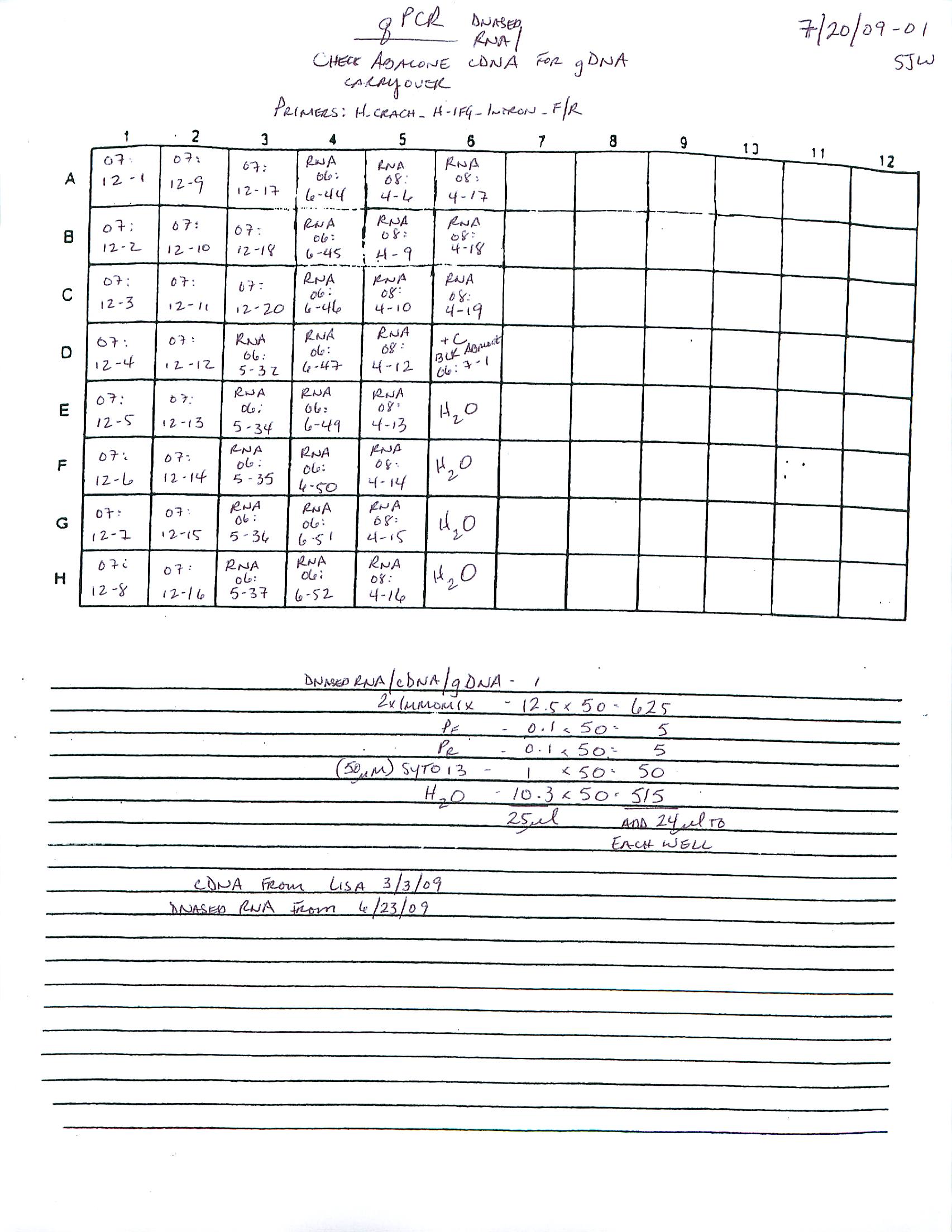{kind=link}
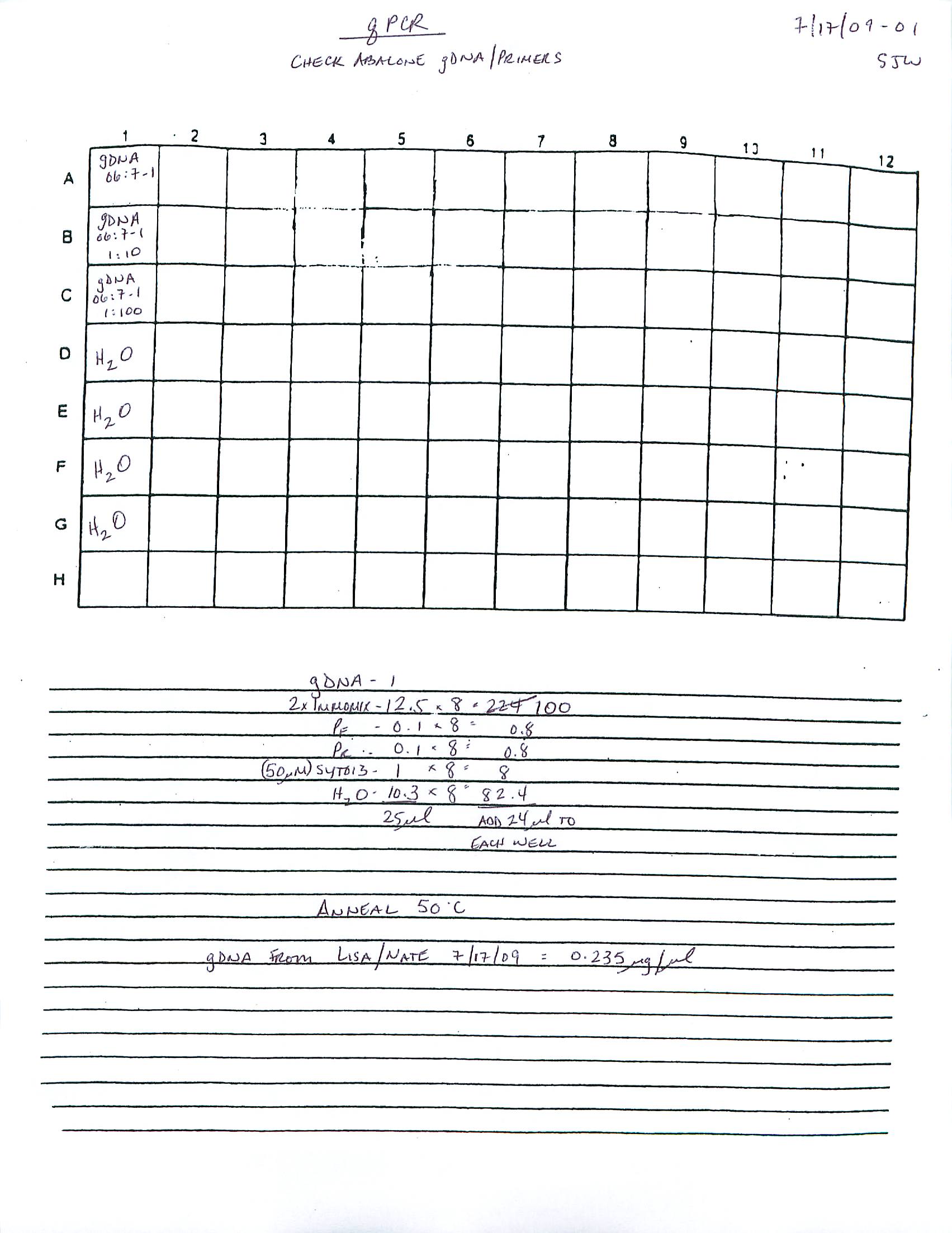{kind=link}
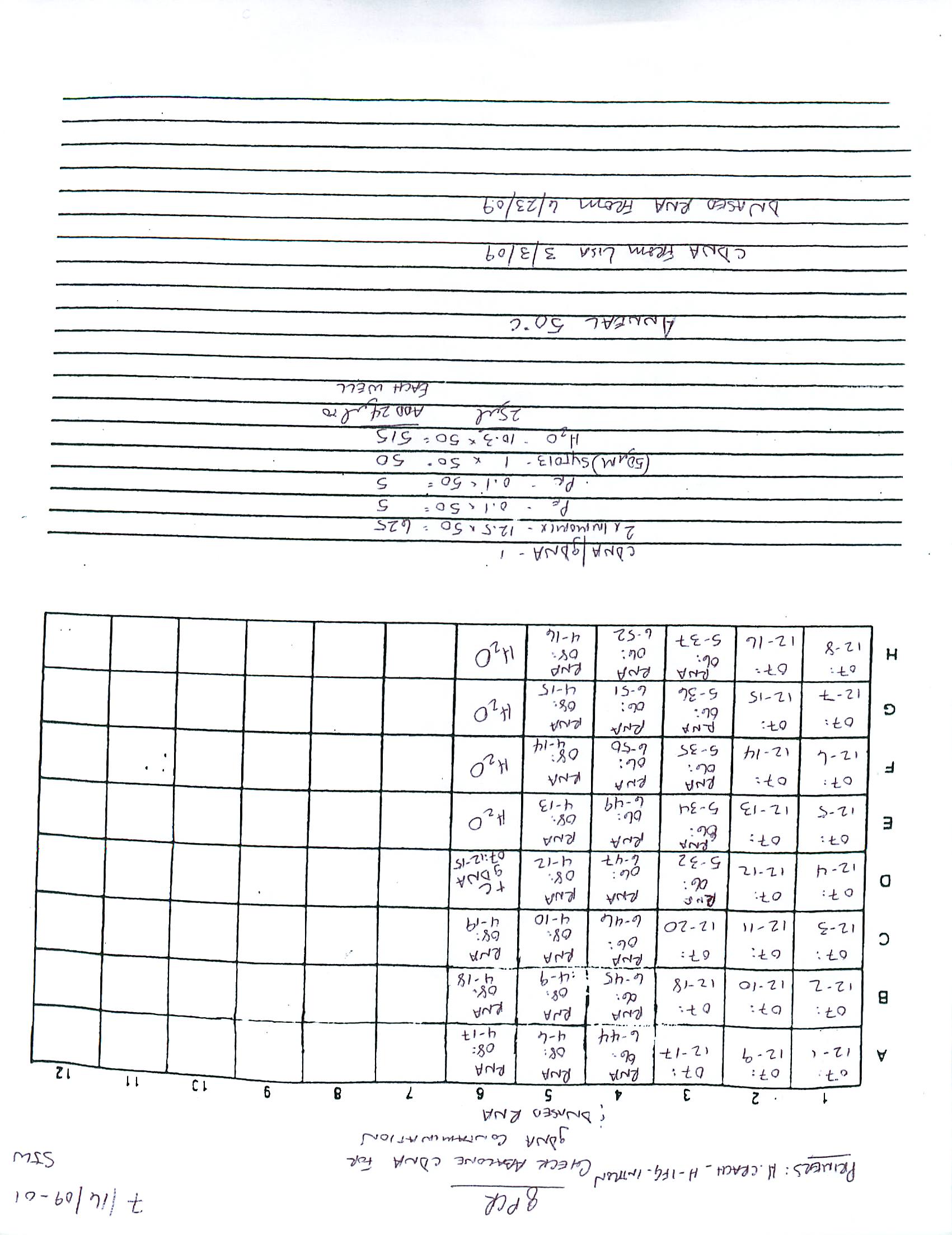{kind=link}
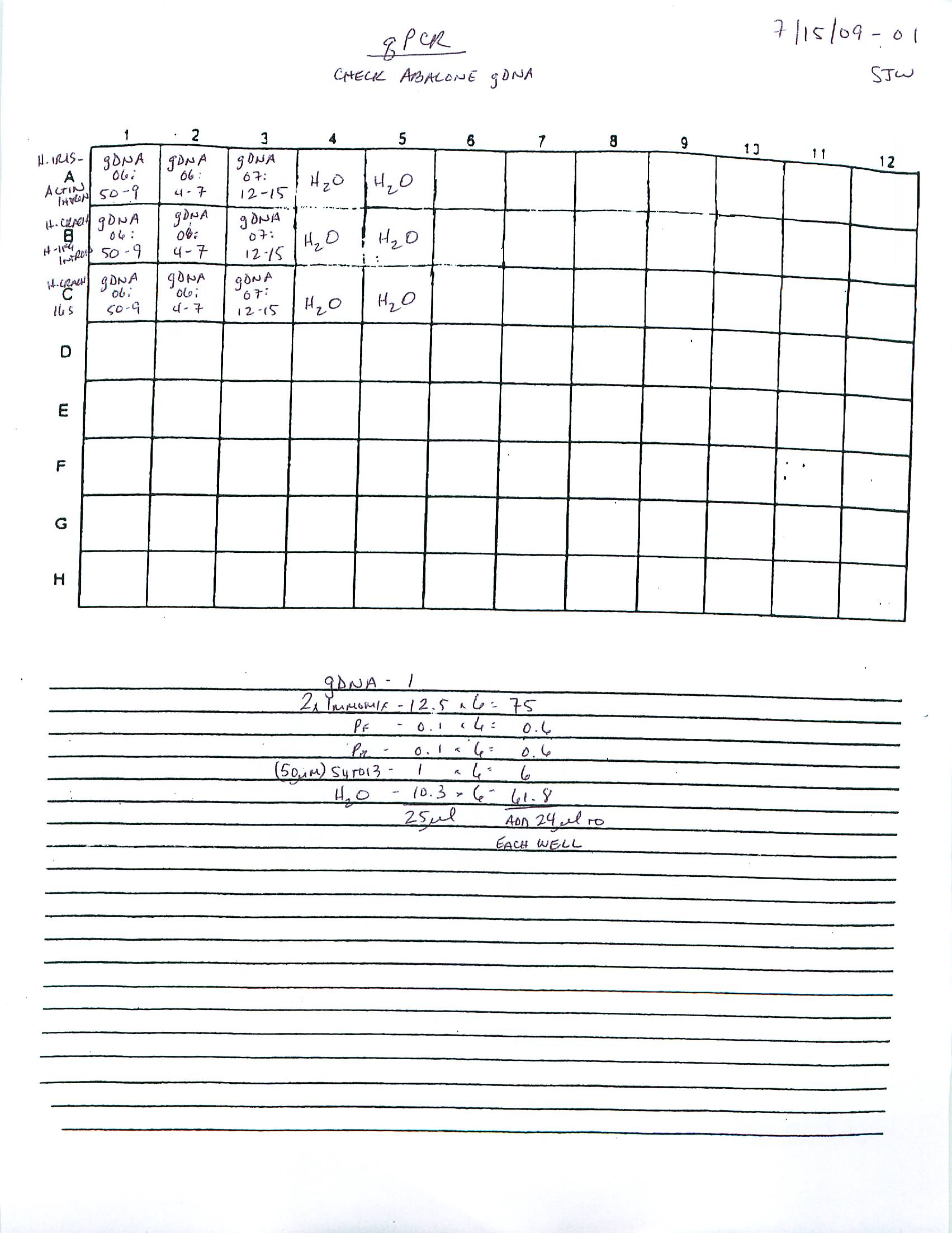{kind=link}
{kind=link}
{kind=link}
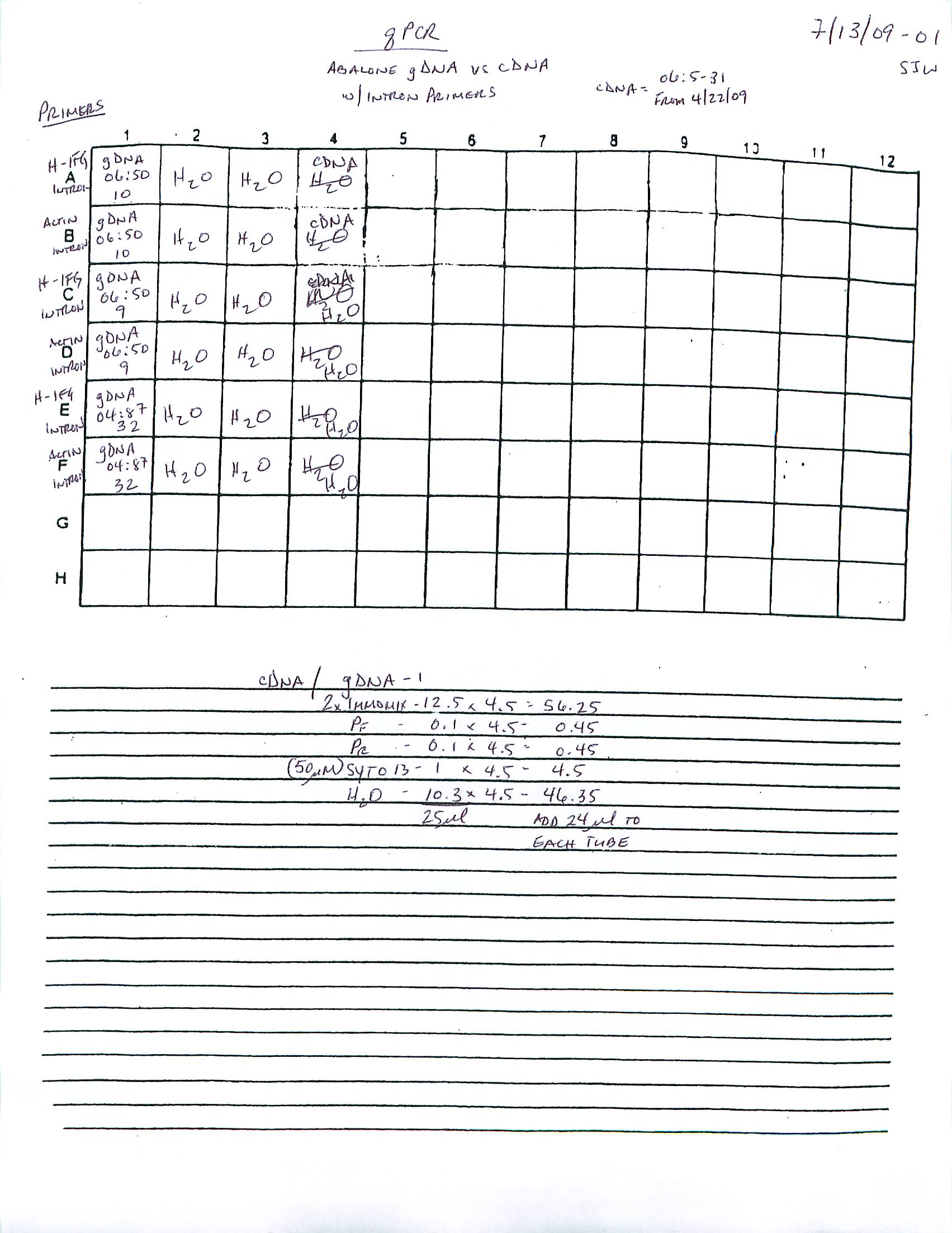{kind=link}
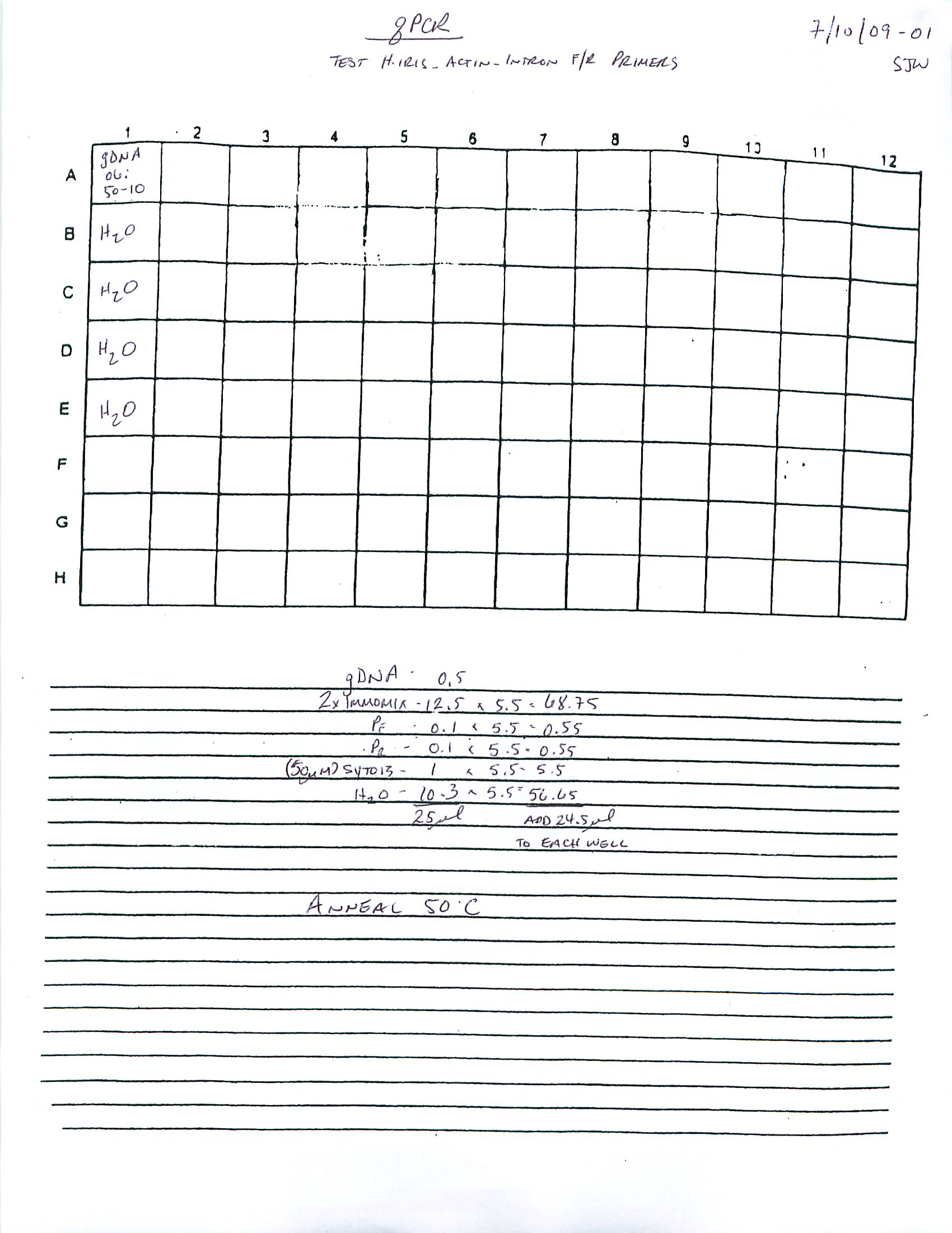{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
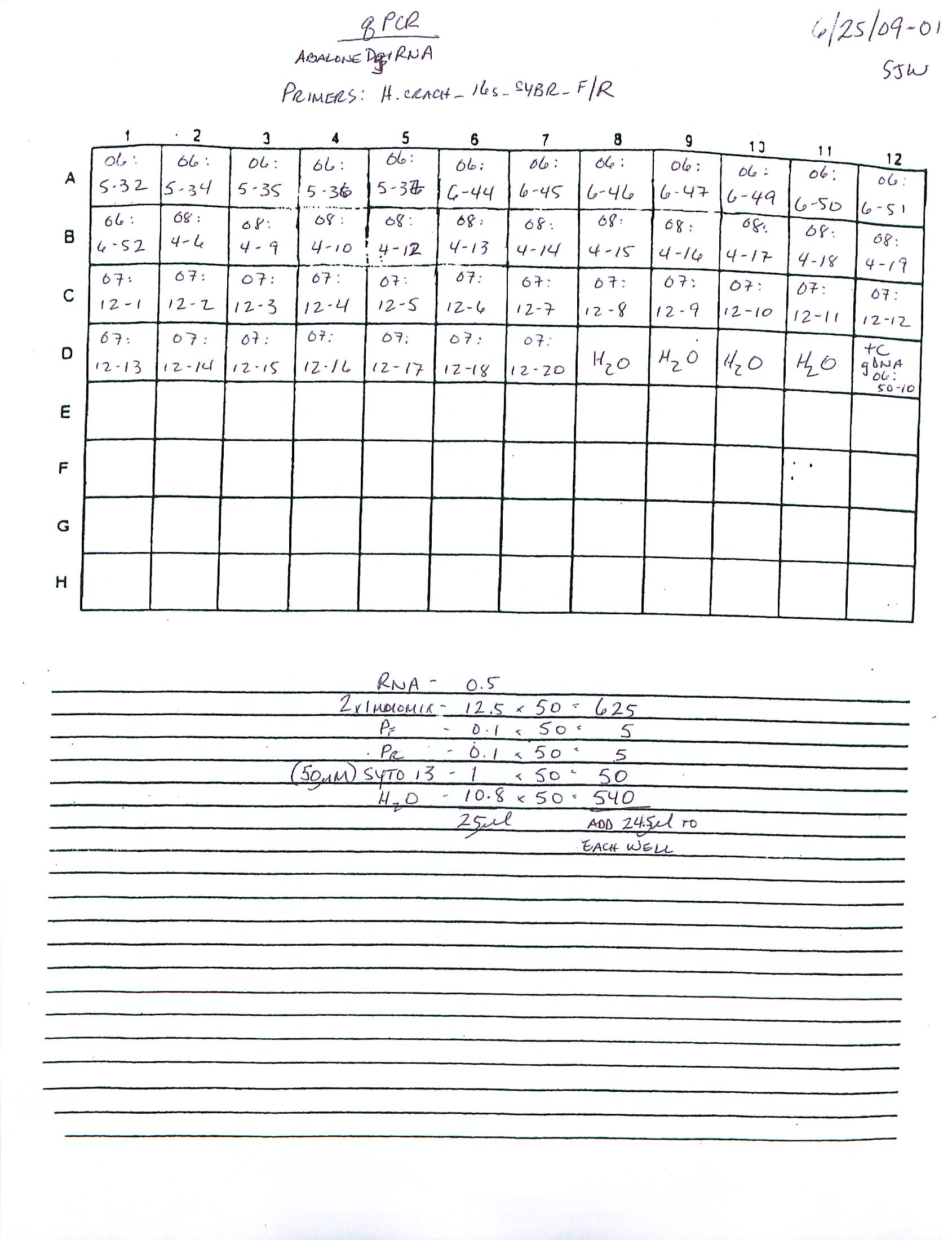{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
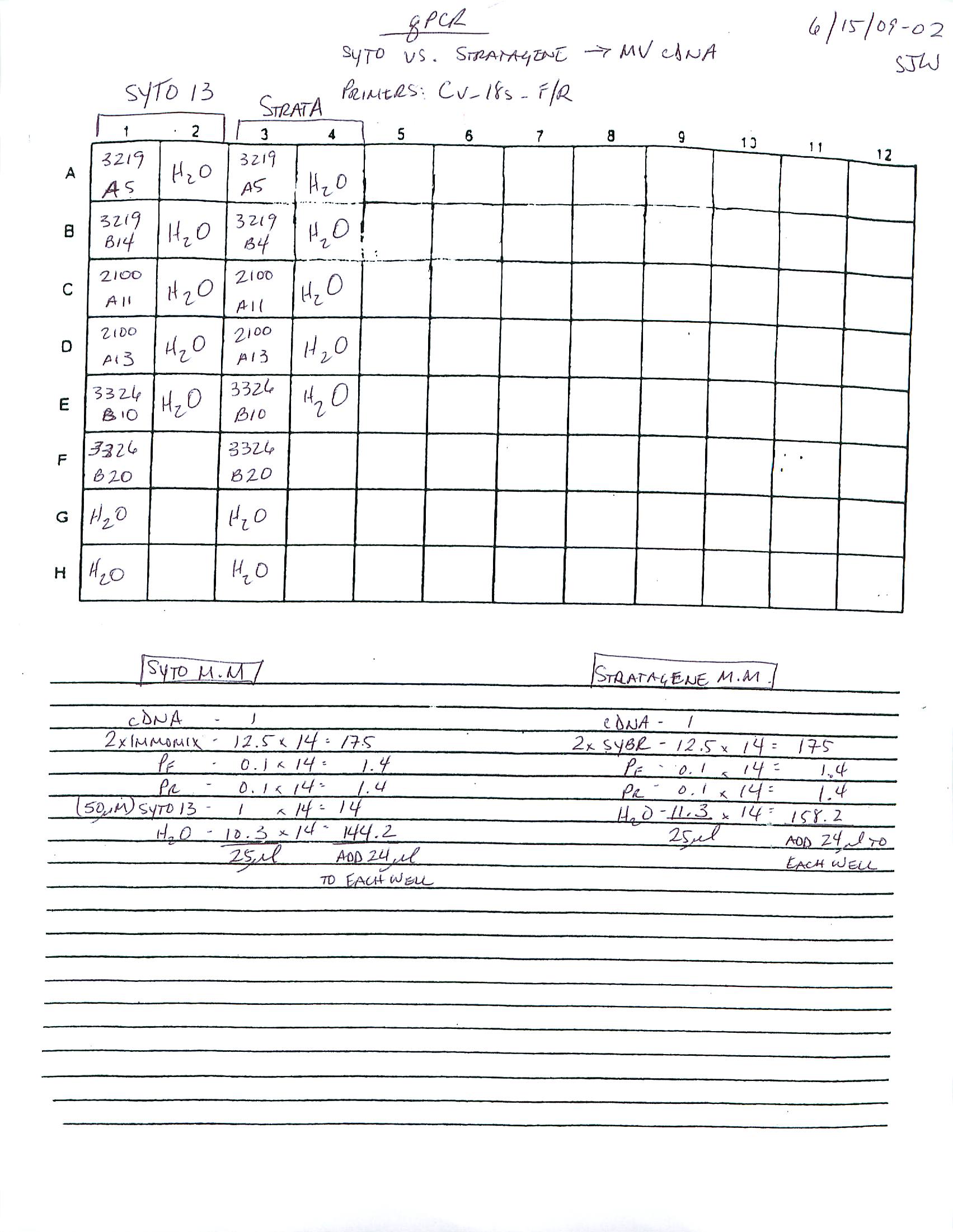{kind=link}
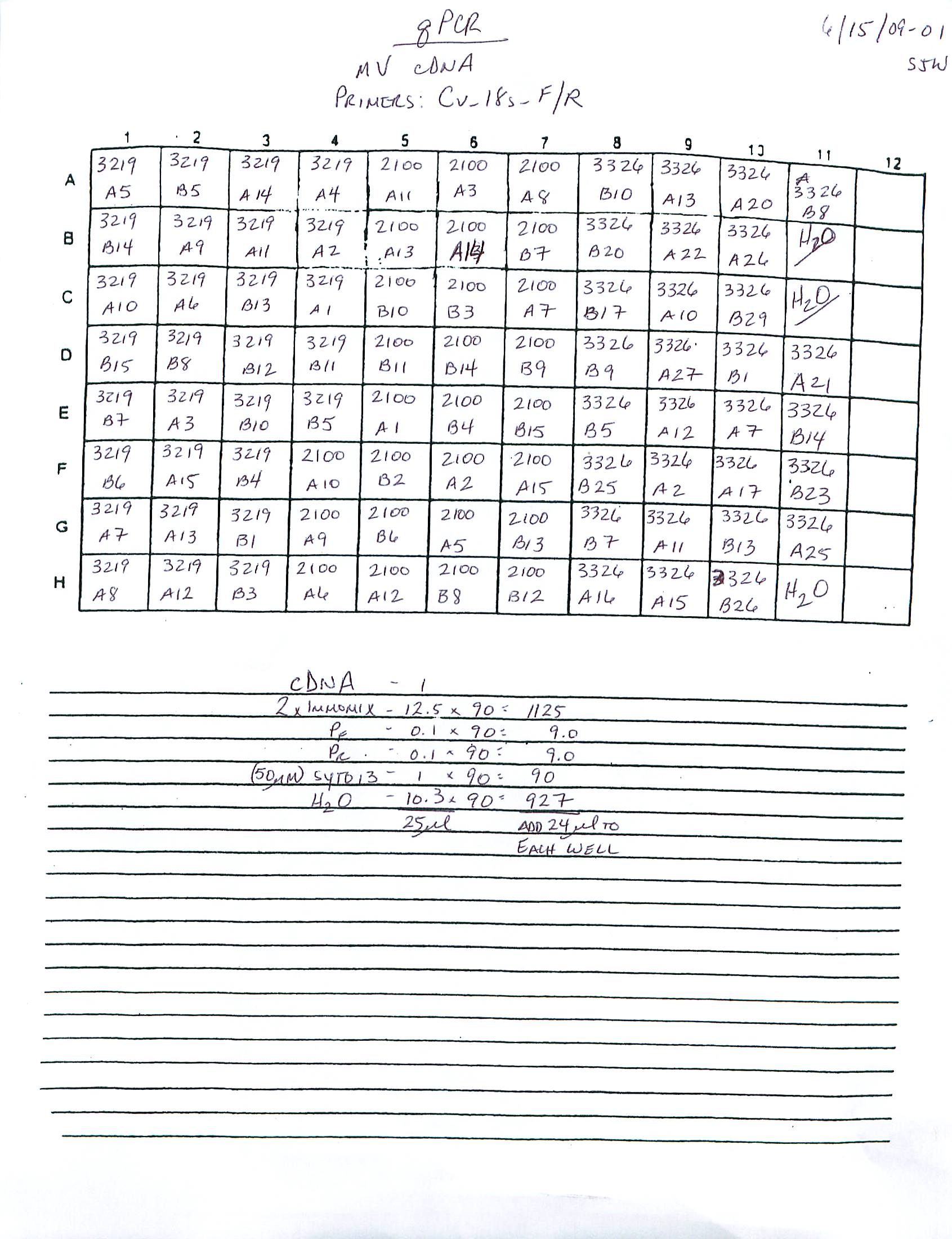{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
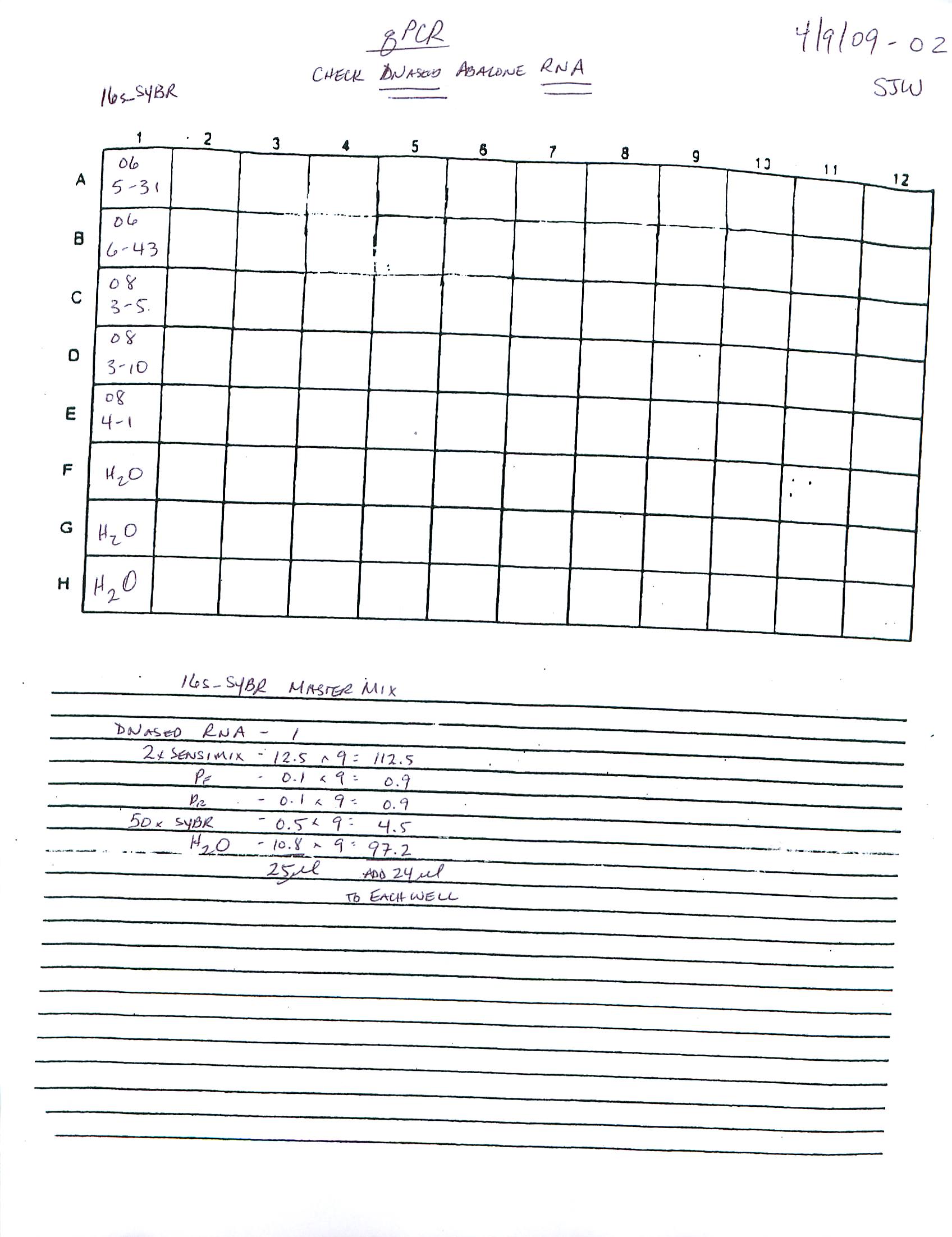{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
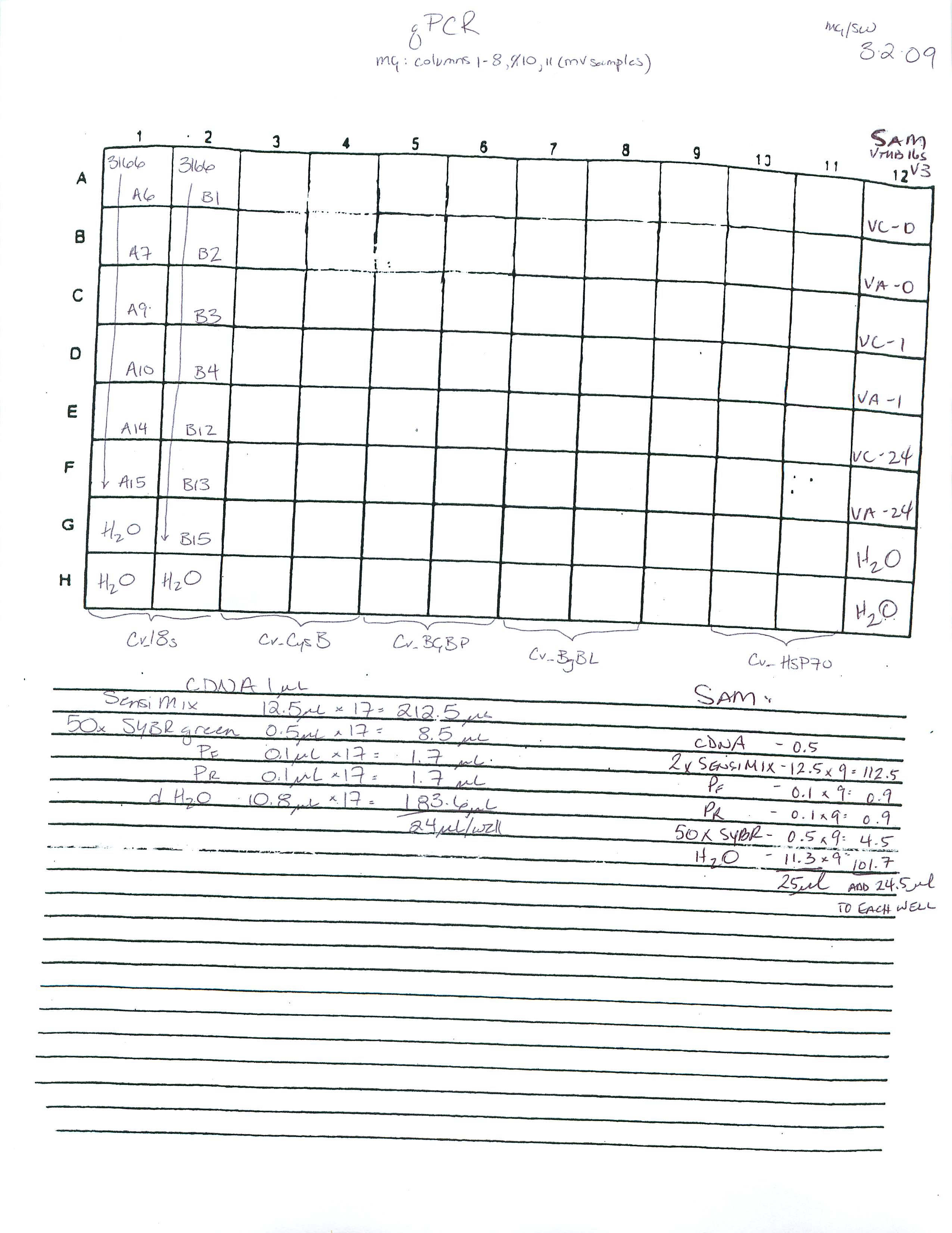{kind=link}
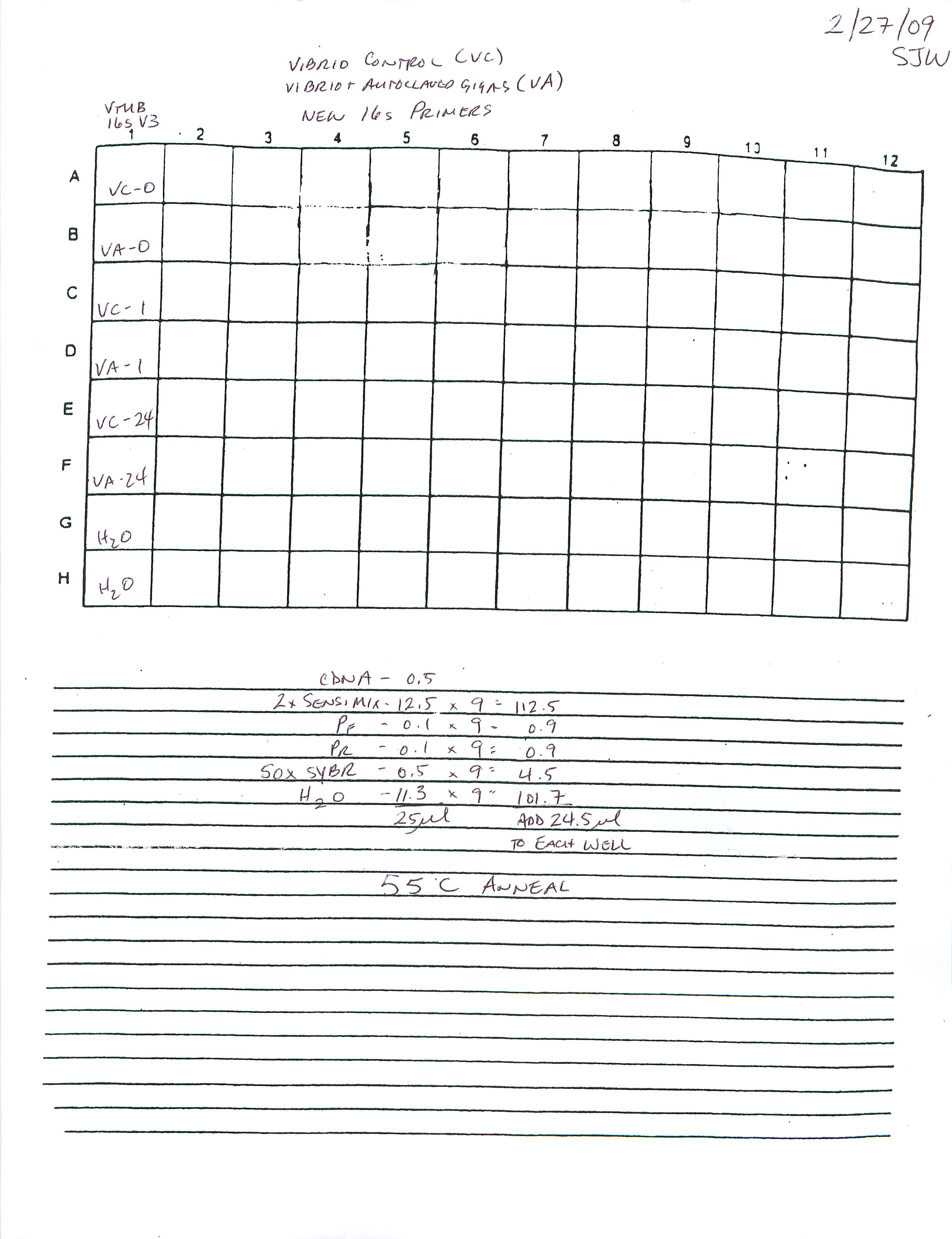{kind=link}
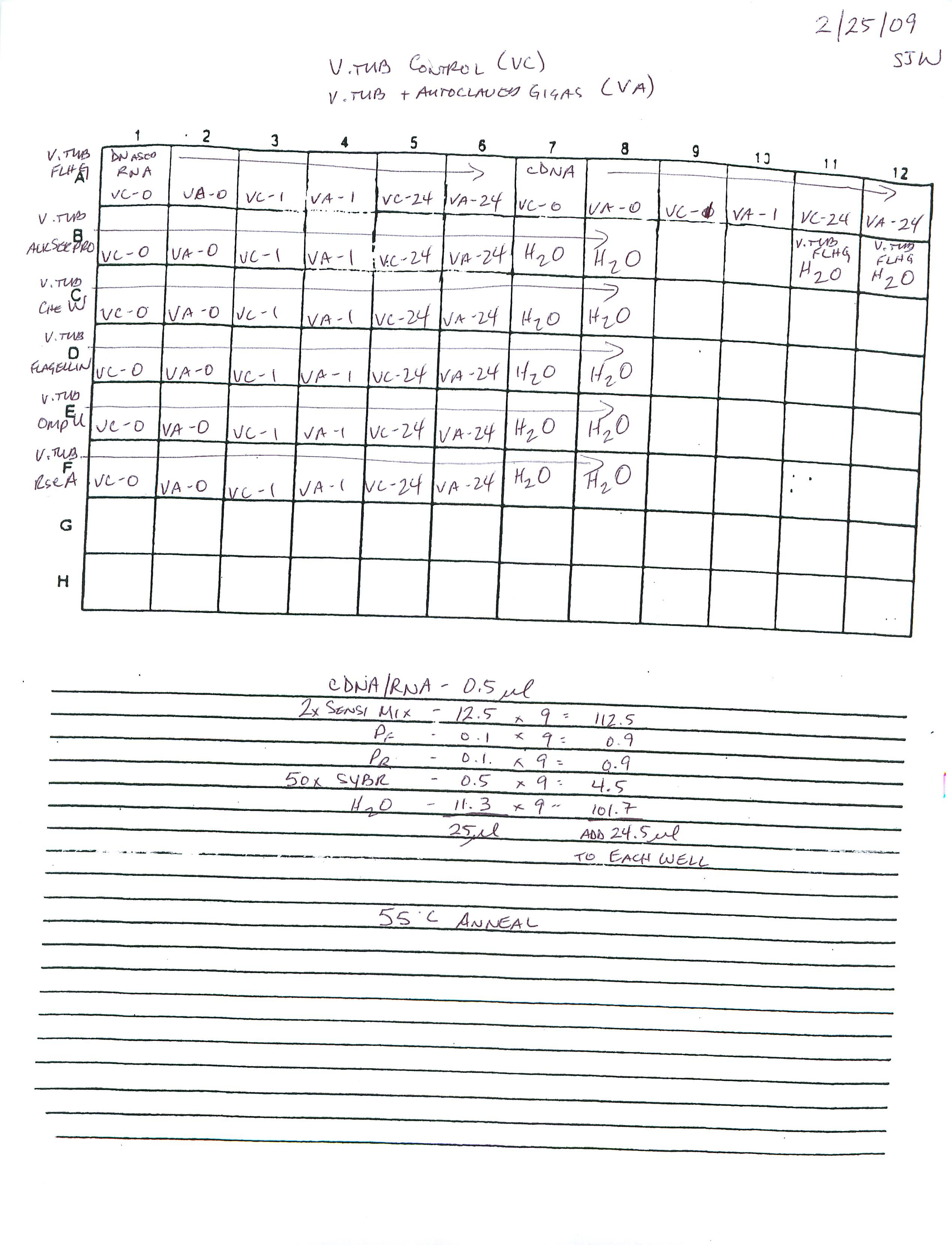{kind=link}
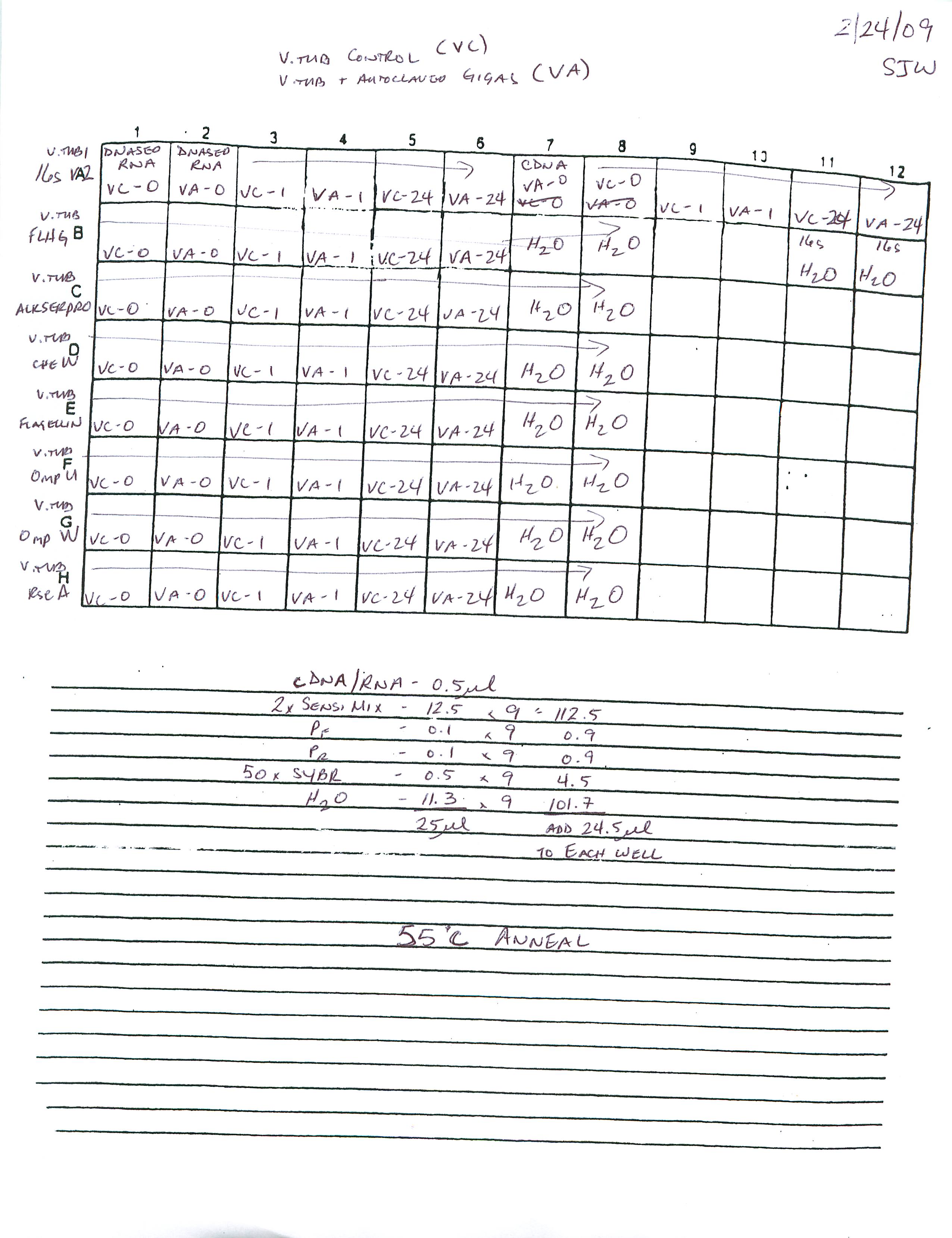{kind=link}
{kind=link}
{kind=link}
rRNA Removal - V. tubiashii total RNA from yesterday
rRNA removal was continued from O/N precipitation. Processed the samples according to the Ambion MICROBExpress Kit protocol and resuspended final pellets in 25uL of The RNA Storage Solution. Samples were spec’d on the NanoDrop.
RNA Isolation - V. tubiashii from challenge (see 20081216)
Added 10mL TriReagent to 2 x 50mL pellets (5.63 x 10^11 total bacteria; see calcs on 20081219) from the control samples collected on 20081219. Added 10mL TriReagent to 1 x 50mL pellet (1.835 x 10^12 total bacteria; see calcs on 20081219) from the V.tubi + oyster samples collected on 20081219. Scaled rest of RNA protocol to match. Resuspended pellets in 100uL 0.1%DEPC-H2O. Samples were NanoDropped.
Vibrio challenge CONTINUED (from yesterday)
Significantly more bacteria in the container containing autoclaved oysters. Collected 2 x 50mL from each treatment. Collected ~750mL from each treatment. Cells were pelleted 4000RPM, 15mins, 4C. Supe was removed and pellets frozen @ -80C.
Vibrio challenge CONTINUED (from yesterday)
500mL culture was split evenly between two containers containing 3L sterile sea water each. One container also contained 3 large, autoclaved C. gigas. Containers had an air stone to promote circulation. 4 x 1mL samples were collected from each container, pelleted @ 10,00RPM 1min. Supe removed and samples stored @ -80C. Samples will be collected @ t = 0, 0.5, 1.0 and 24 hrs. Containers were covered with aluminum foil to minimize splashing caused the by the air stone.
Vibrio challenge CONTINUED (from yesterday)
One of the two starter cultures from yesterday were used to inoculate 500mL 1x LB+1% NaCl and incubated O/N @ 30C 200RPM.
Vibrio challenge
2X 10mL starter cultures of in 1x LB+1% NaCl were inoculated with two separate colonies of V. tubiashii and incubated O/N @ 30C 200RPM.
SDS/PAGE/Western - anti-HSP70 Ab Re-test
Another attempt to determine appropriate amounts of anti-HSP70 Ab [(ABR cat# MA3-006)(https://aquacul4.fish.washington.edu/Protocols:Information%20Sheets/Product%20Information%20Sheets/Antibodies/ABR%20-%20HSP70%20Ab.jpg)and/or protein needed for better detection of HSP70 in Gigas protein samples. Gigas gill protein extracts from 20080617 (both control and Vibrio exposure samples) were each pooled. The two samples were mixed with an equal volume of 2x sample reducing buffer. 100uL of hemolymph were extracted from Gigas muscles and mixed with an equal volume of 2x sample reducing buffer. Samples were boiled for 5mins. and loaded onto a Pierce 4-20% tris-hepes gel. Also loaded 10uL of SeeBlue ladder. Ran gel @ 150V for 45mins. Samples were transferred to nitrocellulose 20V for 30mins. Well locations were marked on the membrane with a pencil. Gel was stained with Coomassie stain for 30 mins and then destained with 10% acetic acid until bands were clearly visible.
SDS/PAGE/Western - Attempt to fix/identify problem(s) with Westerns
Will use two different antibodies and two different development methods: chromogenic (Invitrogen Western Breeze) and chemiluminescent.
SDS/PAGE/Western - anti-HSP70 Ab test CONTINUED (from yesterday)
Primary Ab (anti-HSP70) solution was saved and stored in the 4C. The membrane was washed 3x 10 mins. w/1x TBS-T. 15mL blocking solution was added to membrane and incubated at RT for 10mins. Secondary Ab (DAM HRP) was added at a 1:2500 dilution and inubated at RT for 30 mins. Secondary Ab solution was discarded and membrane was washed 3x 10 mins. w/ 1x TBS-T and then developed with Millipore Immobilon Chemiluminescent reagent.
SDS/PAGE/Western - anti-HSP70 Ab test
Test of the new [anti-HSP70 Ab (ABR cat# MA3-006)(https://aquacul4.fish.washington.edu/Protocols:Information%20Sheets/Product%20Information%20Sheets/Antibodies/ABR%20-%20HSP70%20Ab.jpg). Gigas gill protein extracts from 20080617 (both control and Vibrio exposure samples) were each pooled to result in 10ug protein of control and 10ug protein of VE in a volume of 10uL each. The two samples were mixed with an equal volume of 2x sample reducing buffer. O. rubescans samples were taken from Rachel’s -20C box. 15uL of each sample was mixed with 2x sample reducing buffer. All samples were boiled for 5mins and spot spun. Samples were loaded onto Pierce 4-20% Tris-Glycine SDS/PAGE gels. 10uL of SeeBlue ladder was loaded. Gel was run @ 150V for 45mins.
SDS/PAGE/Western - Purified (His column) FST samples from 20081112
10uL of each sample from 20081112 (binding supe, washes and elution fractions) were mixed with 2x sample reducing buffer and boiled 5 mins. Samples were loaded onto Pierce 4-20% Tris-Glycine SDS/PAGE gels. 10uL of SeeBlue ladder was loaded. 5uL of positive control lysate was loaded. Gel was run @ 150V for 45mins.
Sequencing - Y2H colony PCRs from 20081112
Purified bands were delivered for sequencing. See sequencing log for plate layout/primers.
Mass Spec - Band #1 from 20081106
Trypsin digested band from MSTN1b + red muscle extract co-IP (~75kDa) was delivered for mass spec analysis. Data in 2-3 weeks.
Western Blot - Purified (His column) decorin, FST, LAP & telethonin
10uL of each sample from yesterday (binding supe, washes and elution fractions) were mixed with 2x sample reducing buffer and boiled 5 mins. Samples were loaded onto Pierce 4-20% Tris-Glycine SDS/PAGE gels. 10uL of SeeBlue ladder was loaded. 5uL of positive control lysate was loaded. Gels were run @ 150V for 45mins.