INTRO
Steven asked me to perform resazurin assays on “selected” juvenile Ruditapes philippinarum (Manila clam) exposed to 36oC acute heat stress. The goal of the experiment is to compare metabolic activity between the “selected” clams and control clams when exposed to heat stress. This is similar to the previous experiment at 36oC Day 0 Notebook Entry and Day 1 Notebook Entry, but with a set of larger clams.
Data and results are available here:
The section below was knitted from the R Markdown file 00.00-resazurin-20260401-clam-36C-C-vs-S.Rmd (GitHub).
1 Background
Control clams (C) compared with selected clams (S) at 36°C using a resazurin metabolic assay. Clams were placed in 12-well plates submerged in 5.0 mL resazurin working solution (prepared 2026-03-30 by SJW, stored at 4°C). Plate layout was randomised (treatment assignments known only to Steven Roberts). Fluorescence was measured every 30 min on a Synergy HTX (Agilent) plate reader.
Important notes from this experiment:
- The following wells showed low volume and are flagged for exclusion in the layout: G A3, J B1, K A2, L A4.
- Plate G was slow to reach target temperature: wells ranged 11–12°C at T0, reaching 33°C only by T4. Interpret Plate G data with caution.
- Plate G T0 was accidentally scanned as a 24-well plate; the plate consistency check will detect and exclude Plate G automatically.
See data/clam/20260401-clam-36C-C-vs-S/README.md for full experimental notes including per-timepoint temperature spot checks.
1.1 Expected inputs
| Path | Description |
|---|---|
data/clam/20260401-clam-36C-C-vs-S/plate-*-T*.txt |
Plate reader fluorescence exports (one file per plate per timepoint) |
data/clam/20260401-clam-36C-C-vs-S/layout.csv |
Well metadata: plate ID, well ID, blank flag, family/treatment groups, size measurements, exclusion flags |
1.2 Expected outputs
All outputs are written to output/clam/20260401-clam-36C-C-vs-S/.
| File | Description |
|---|---|
figures/ |
All plots generated by this script |
auc_all_metrics.csv |
Per-individual AUC values for every active measurement metric |
auc_summary.csv |
Group-level AUC summary statistics (mean, SD, SE, median) |
metabolism.csv |
Full per-well per-timepoint metabolism data frame |
pairwise_stats.csv |
Tukey-adjusted pairwise comparisons from AUC linear models |
2 Setup
knitr::opts_chunk$set(
echo = TRUE, # Display code chunks
eval = TRUE, # Evaluate code chunks
warning = FALSE, # Hide warnings
message = FALSE, # Hide messages
comment = "", # Prevents appending '##' to beginning of lines in code output
results = 'hold' # Holds output so it's all printed together after code chunk
)library(tidyverse)
library(pracma) # trapz()
library(lme4)
library(lmerTest)
library(emmeans)
library(multcompView)
library(cowplot)
library(colorspace) # qualitative_hcl() for large palettes3 Helper Functions
normalize_well_id <- function(x) {
x <- toupper(trimws(x))
valid <- str_detect(x, "^[A-Z]+[0-9]+$")
out <- rep(NA_character_, length(x))
if (!any(valid)) return(out)
m <- str_match(x[valid], "^([A-Z]+)([0-9]+)$")
out[valid] <- paste0(m[, 2], as.integer(m[, 3]))
out
}
parse_time_hr <- function(path) {
hit <- str_match(basename(path),
"(?i)-T([0-9]+(?:\\.[0-9]+)?)\\.txt$")
as.numeric(hit[, 2])
}
parse_plate_id <- function(path) {
hit <- str_match(basename(path),
"(?i)^plate-([A-Za-z0-9-]+)-T[0-9]+(?:\\.[0-9]+)?\\.txt$")
id <- hit[, 2]
ifelse(is.na(id), "unknown", id)
}
extract_results_block <- function(lines) {
results_idx <- which(trimws(lines) == "Results")
if (length(results_idx) == 0) stop("No Results section found")
idx <- results_idx[1]
header_tokens <- str_split(lines[idx + 1], "\\t")[[1]] |> trimws()
col_ids <- header_tokens[
header_tokens != "" & str_detect(header_tokens, "^[0-9]+$")]
j <- idx + 2
data_lines <- character()
while (j <= length(lines)) {
line <- lines[j]
if (trimws(line) == "") break
if (!str_detect(line, "^[A-Za-z]\\t")) break
data_lines <- c(data_lines, line)
j <- j + 1
}
list(col_ids = col_ids, data_lines = data_lines)
}
parse_plate_export <- function(path) {
lines <- readLines(path, warn = FALSE)
res <- extract_results_block(lines)
map_dfr(res$data_lines, function(line) {
tokens <- str_split(line, "\\t")[[1]] |> trimws()
tokens <- tokens[tokens != ""]
row_letter <- tokens[1]
nums <- suppressWarnings(as.numeric(tokens[-1]))
valid_idx <- which(!is.na(nums))
if (length(valid_idx) == 0) return(tibble())
vals <- nums[valid_idx]
n <- min(length(vals), length(res$col_ids))
tibble(
row_id = toupper(row_letter),
col_id = as.integer(res$col_ids[seq_len(n)]),
well_id = normalize_well_id(
paste0(toupper(row_letter), res$col_ids[seq_len(n)])),
value = vals[seq_len(n)]
)
}) %>%
mutate(
plate_id = str_to_lower(parse_plate_id(path)),
time_hr = parse_time_hr(path)
)
}
trapezoid_auc <- function(time_hr, value) {
ok <- is.finite(time_hr) & is.finite(value)
t <- time_hr[ok]
v <- value[ok]
if (length(t) < 2) return(NA_real_)
ord <- order(t)
t <- t[ord]; v <- v[ord]
sum(diff(t) * (head(v, -1) + tail(v, -1)) / 2)
}
# Shared helper: extract display unit string from a measurement column name.
# e.g. "area_mm2_measurement" -> "mm²", "weight_mg_measurement" -> "mg"
parse_meas_unit <- function(col_name) {
unit_raw <- col_name |>
str_remove("^metabolism_per_") |>
str_remove("_measurement$") |>
str_extract("[^_]+$")
case_when(
unit_raw == "mm2" ~ "mm²",
unit_raw == "cm2" ~ "cm²",
unit_raw == "mm3" ~ "mm³",
unit_raw == "cm3" ~ "cm³",
TRUE ~ unit_raw
)
}
# y-axis label for metabolism line plots: "fold change/mm²"
metabolism_y_label <- function(col_name) {
paste0("Metabolism (fold change/", parse_meas_unit(col_name), ")")
}
# y-axis label for AUC box plots: "Metabolism (AUC; mm²)"
auc_y_label <- function(metric_name) {
paste0("Metabolism (AUC; ", parse_meas_unit(metric_name), ")")
}4 Load Data
4.1 Plate export files
proj_root <- rprojroot::find_rstudio_root_file()
data_dir <- file.path(proj_root, "data", "clam",
"20260401-clam-36C-C-vs-S")
fig_dir <- file.path(proj_root, "output", "clam",
"20260401-clam-36C-C-vs-S", "figures")
out_dir <- file.path(proj_root, "output", "clam",
"20260401-clam-36C-C-vs-S")
dir.create(fig_dir, recursive = TRUE, showWarnings = FALSE)
dir.create(out_dir, recursive = TRUE, showWarnings = FALSE)
plate_files <- list.files(
data_dir,
pattern = "(?i)^plate-.*-T[0-9]+(?:\\.[0-9]+)?\\.txt$",
full.names = TRUE
)
plate_raw <- map_dfr(plate_files, function(path) {
tryCatch(parse_plate_export(path),
error = function(e) {
message("Parse error in ", basename(path), ": ", e$message)
tibble()
})
})
str(plate_raw)tibble [660 × 6] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:660] "A" "A" "A" "A" ...
$ col_id : int [1:660] 1 2 3 4 5 6 1 2 3 4 ...
$ well_id : chr [1:660] "A1" "A2" "A3" "A4" ...
$ value : num [1:660] 350 75 417 326 303 360 122 2 255 353 ...
$ plate_id: chr [1:660] "g" "g" "g" "g" ...
$ time_hr : num [1:660] 0 0 0 0 0 0 0 0 0 0 ...4.2 Plate consistency check
Checks that every plate has the same number of wells at every timepoint. The expected well count is the mode across all plate × timepoint reads. Any plate with at least one deviating read is flagged and dropped entirely before any further analysis — removing only the aberrant timepoint would break the fold-change baseline calculation.
well_counts <- plate_raw %>%
group_by(plate_id, time_hr) %>%
summarise(n_wells = n_distinct(well_id), .groups = "drop")
expected_n_wells <- as.integer(
names(which.max(table(well_counts$n_wells)))
)
inconsistent_reads <- well_counts %>%
filter(n_wells != expected_n_wells) %>%
arrange(plate_id, time_hr)
inconsistent_plate_ids <- unique(inconsistent_reads$plate_id)
if (nrow(inconsistent_reads) > 0) {
cat("**Plate consistency check FAILED.**",
"Expected", expected_n_wells, "wells per plate-timepoint read.",
length(inconsistent_plate_ids),
"plate(s) have at least one deviating read and are excluded",
"from all analyses:\n\n")
cat(knitr::kable(
inconsistent_reads,
col.names = c("Plate", "Time (h)", "Wells read"),
caption = paste("Expected:", expected_n_wells, "wells per read")
), sep = "\n")
cat("\n")
plate_raw <- plate_raw %>%
filter(!plate_id %in% inconsistent_plate_ids)
message(length(inconsistent_plate_ids),
" plate(s) removed from plate_raw: ",
paste(inconsistent_plate_ids, collapse = ", "))
} else {
cat("Plate consistency check passed: all",
n_distinct(well_counts$plate_id), "plates have",
expected_n_wells, "wells at every timepoint.\n")
}Plate consistency check FAILED. Expected 12 wells per plate-timepoint read. 1 plate(s) have at least one deviating read and are excluded from all analyses:
| Plate | Time (h) | Wells read |
|---|---|---|
| g | 0 | 24 |
Expected: 12 wells per read
4.3 Layout file
layout_path <- file.path(data_dir, "layout.csv")
layout_raw <- read_csv(layout_path,
col_types = cols(.default = "c"),
show_col_types = FALSE)
# Standardise column names to snake_case
names(layout_raw) <- names(layout_raw) |>
str_to_lower() |>
str_replace_all("[^a-z0-9]+", "_") |>
str_replace_all("_+", "_") |>
str_replace("_$", "")
# Normalise plate_id to match plate file ids (strip "plate-" prefix)
layout_clean <- layout_raw %>%
mutate(
plate_id = str_remove(str_to_lower(plate_id), "^plate-"),
well_id = normalize_well_id(plate_well),
is_blank = if ("is_blank" %in% names(layout_raw))
toupper(trimws(is_blank)) %in% c("TRUE", "T", "1", "YES", "Y")
else
FALSE
)
found_exclude_col <- intersect(
c("exclude_from_analysis", "exclude", "omit", "not_analyzed"),
names(layout_clean)
)[1]
layout_clean <- layout_clean %>%
mutate(
exclude_from_analysis = if (!is.na(found_exclude_col))
toupper(trimws(.data[[found_exclude_col]])) %in%
c("TRUE", "T", "1", "YES", "Y")
else
FALSE
)
# Identify measurement columns and group columns
measurement_cols <- names(layout_clean)[
str_detect(names(layout_clean), "_measurement$")]
group_cols <- names(layout_clean)[
str_detect(names(layout_clean), "_group$")]
# Cast measurement columns to numeric
layout_clean <- layout_clean %>%
mutate(across(all_of(measurement_cols),
~ suppressWarnings(as.numeric(.x))))
# Determine which measurement columns actually contain finite data
active_meas_cols <- measurement_cols[
sapply(measurement_cols, function(col)
any(is.finite(layout_clean[[col]]), na.rm = TRUE))]
# Normalise group values to lowercase so they match colour scale definitions
layout_clean <- layout_clean %>%
mutate(across(all_of(group_cols),
~ str_to_lower(trimws(as.character(.x)))))
message("Group columns: ", paste(group_cols, collapse = ", "))
message("Active measurement columns: ",
paste(active_meas_cols, collapse = ", "))
str(layout_clean)tibble [72 × 14] (S3: tbl_df/tbl/data.frame)
$ plate_id : chr [1:72] "g" "g" "g" "g" ...
$ plate_well : chr [1:72] "A01" "A02" "A03" "A04" ...
$ is_blank : logi [1:72] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ family_id_group : chr [1:72] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:72] "1" "2" "3" "4" ...
$ treatment_group : chr [1:72] "selected" "selected" "control" "control" ...
$ exclude_from_analysis: logi [1:72] FALSE FALSE TRUE FALSE FALSE FALSE ...
$ exclude_reason : chr [1:72] NA NA "volume loss" NA ...
$ width_mm_measurement : num [1:72] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:72] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:72] NA NA NA NA NA NA NA NA NA NA ...
$ area_cm2_measurement : num [1:72] 1.585 1.311 1.83 0.818 1.274 ...
$ imagej_id : chr [1:72] "3" "4" "1" "2" ...
$ well_id : chr [1:72] "A1" "A2" "A3" "A4" ...5 Merge Plate Data with Layout
dat <- plate_raw %>%
left_join(
layout_clean %>%
select(plate_id, well_id, is_blank, exclude_from_analysis,
any_of("exclude_reason"),
all_of(group_cols), all_of(measurement_cols)),
by = c("plate_id", "well_id")
) %>%
mutate(
is_blank = replace_na(is_blank, FALSE),
exclude_from_analysis = replace_na(exclude_from_analysis, FALSE)
)
str(dat)tibble [540 × 16] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:540] "A" "A" "A" "A" ...
$ col_id : int [1:540] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:540] "A1" "A2" "A3" "A4" ...
$ value : num [1:540] 368 350 302 310 316 309 256 336 318 201 ...
$ plate_id : chr [1:540] "h" "h" "h" "h" ...
$ time_hr : num [1:540] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:540] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:540] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_reason : chr [1:540] NA NA NA NA ...
$ family_id_group : chr [1:540] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:540] "13" "14" "15" "16" ...
$ treatment_group : chr [1:540] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:540] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:540] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:540] NA NA NA NA NA NA NA NA NA NA ...
$ area_cm2_measurement : num [1:540] 1.57 1.31 1.1 1.08 1.23 ...6 Raw Fluorescence
6.1 Data frame
# Wells in the plate reader output that have no layout entry get all-NA group
# columns after the join. Keep only wells assigned to at least one group.
active_gc <- intersect(group_cols, names(dat))
raw_df <- dat %>%
filter(
!is_blank,
if (length(active_gc) > 0)
if_any(all_of(active_gc), ~ !is.na(.))
else
TRUE
) %>%
mutate(
trace_id = if_else(
!is.na(sample_id_group) & trimws(as.character(sample_id_group)) != "",
as.character(sample_id_group),
paste(plate_id, well_id, sep = "_")
)
)
families <- sort(unique(na.omit(raw_df$family_id_group)))
treatments <- sort(unique(na.omit(raw_df$treatment_group)))
n_fam <- length(families)
n_trt <- length(treatments)
# Palette strategy:
# <= 7 groups : Okabe-Ito (gold standard for colorblind-safe figures).
# > 7 groups : colorspace::qualitative_hcl("Dynamic") scales to any N
# using perceptually uniform HCL space — no colour collisions.
# Black (#000000) is excluded from both and reserved for blank wells.
okabe_ito_7 <- c(
"#E69F00", "#56B4E9", "#009E73", "#F0E442",
"#0072B2", "#D55E00", "#CC79A7"
)
make_palette <- function(n) {
if (n == 0L) return(character(0))
if (n <= length(okabe_ito_7)) return(okabe_ito_7[seq_len(n)])
colorspace::qualitative_hcl(n, palette = "Dynamic")
}
all_colours <- make_palette(n_fam + n_trt)
fam_colours <- setNames(all_colours[seq_len(n_fam)], families)
trt_colours <- setNames(all_colours[n_fam + seq_len(n_trt)], treatments)
lty_pool <- c("solid", "dashed", "dotted", "dotdash", "longdash")
trt_linetypes <- setNames(
lty_pool[(seq_len(n_trt) - 1L) %% length(lty_pool) + 1L],
treatments
)
plate_well_colours <- c(blank = "black", fam_colours)
str(raw_df)tibble [495 × 17] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:495] "A" "A" "A" "A" ...
$ col_id : int [1:495] 1 2 3 4 1 2 3 4 1 3 ...
$ well_id : chr [1:495] "A1" "A2" "A3" "A4" ...
$ value : num [1:495] 368 350 302 310 316 309 256 336 318 317 ...
$ plate_id : chr [1:495] "h" "h" "h" "h" ...
$ time_hr : num [1:495] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:495] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:495] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_reason : chr [1:495] NA NA NA NA ...
$ family_id_group : chr [1:495] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:495] "13" "14" "15" "16" ...
$ treatment_group : chr [1:495] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:495] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:495] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:495] NA NA NA NA NA NA NA NA NA NA ...
$ area_cm2_measurement : num [1:495] 1.57 1.31 1.1 1.08 1.23 ...
$ trace_id : chr [1:495] "13" "14" "15" "16" ...6.2 Raw fluorescence by plate (including blanks)
p_raw_plates <- dat %>%
filter(is.finite(time_hr), is.finite(value)) %>%
mutate(
colour_group = if_else(is_blank, "blank",
coalesce(family_id_group, "sample")),
trace_id = paste(plate_id, well_id, sep = "_")
) %>%
ggplot(aes(x = time_hr, y = value,
group = trace_id, colour = colour_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1, alpha = 0.7) +
facet_wrap(~ plate_id) +
scale_colour_manual(
values = plate_well_colours,
name = "Group",
breaks = names(plate_well_colours),
na.value = "grey80"
) +
labs(x = "Time (h)", y = "Raw fluorescence (RFU)") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_raw_plates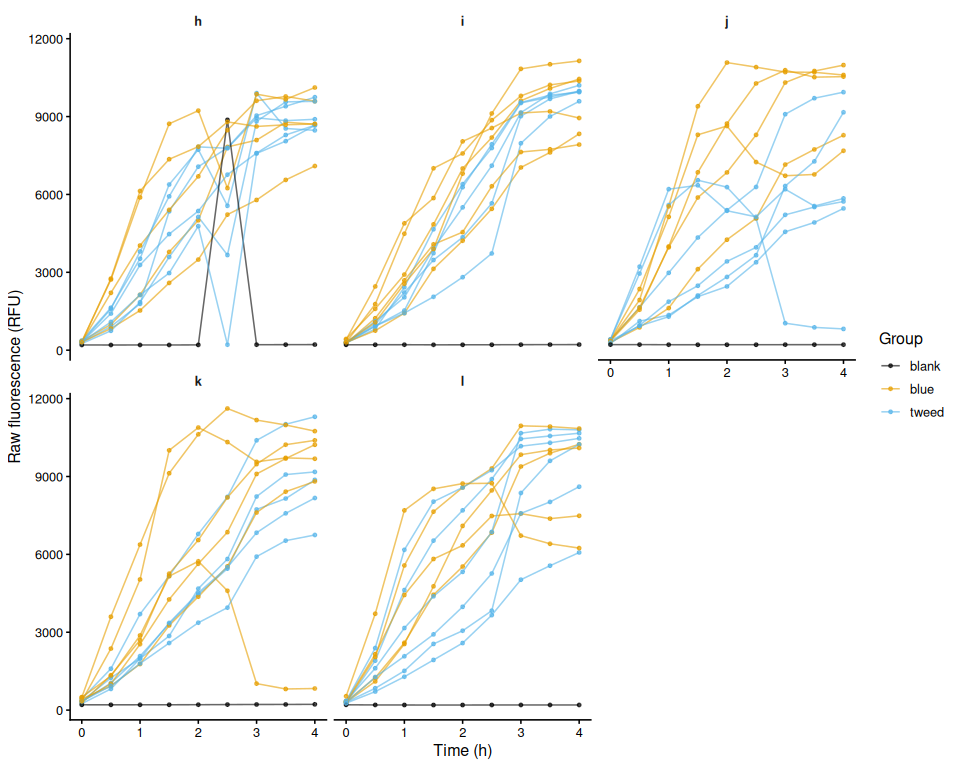
ggsave(file.path(fig_dir, "raw_fluor_by_plate.png"),
p_raw_plates, width = 10, height = 8)6.3 Mean raw fluorescence by family
raw_family_summary <- raw_df %>%
group_by(family_id_group, treatment_group, time_hr) %>%
summarise(
mean_fluor = mean(value, na.rm = TRUE),
se_fluor = sd(value, na.rm = TRUE) /
sqrt(sum(!is.na(value))),
n = sum(!is.na(value)),
.groups = "drop"
)
p_raw_mean <- ggplot(raw_family_summary,
aes(x = time_hr, y = mean_fluor,
colour = family_id_group, linetype = treatment_group,
group = interaction(family_id_group, treatment_group))) +
geom_ribbon(aes(ymin = mean_fluor - se_fluor,
ymax = mean_fluor + se_fluor,
fill = family_id_group),
alpha = 0.15, colour = NA) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_colour_manual(values = fam_colours, name = "Family") +
scale_fill_manual(values = fam_colours, name = "Family") +
scale_linetype_manual(values = trt_linetypes, name = "Treatment") +
labs(x = "Time (h)", y = "Mean raw fluorescence (RFU ± SE)") +
theme_classic(base_size = 13)
p_raw_mean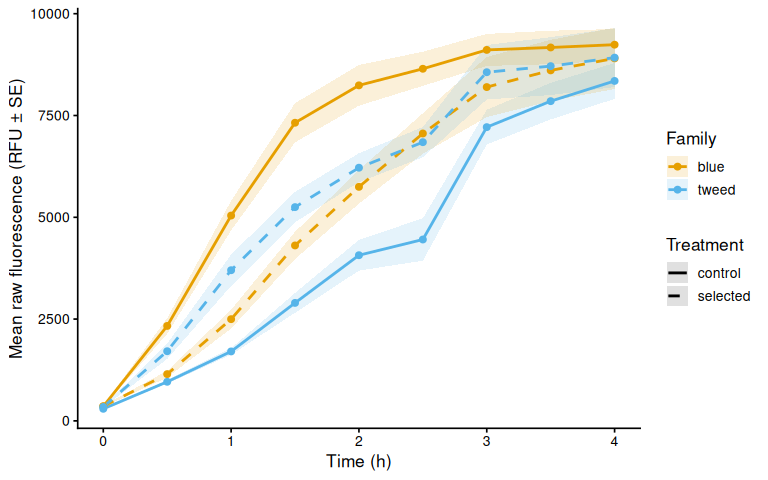
ggsave(file.path(fig_dir, "raw_mean_by_family.png"),
p_raw_mean, width = 8, height = 5)6.4 Individual raw fluorescence traces by family
p_raw_by_family <- raw_df %>%
ggplot(aes(x = time_hr, y = value,
group = trace_id, colour = treatment_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ family_id_group) +
scale_colour_manual(values = trt_colours, name = "Treatment") +
labs(x = "Time (h)", y = "Raw fluorescence (RFU)") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_raw_by_family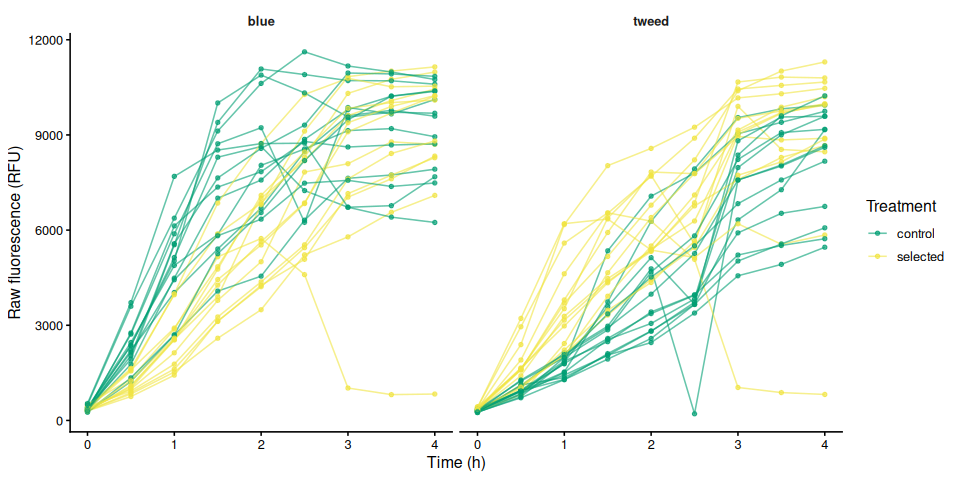
ggsave(file.path(fig_dir, "raw_individual_by_family.png"),
p_raw_by_family, width = 10, height = 5)6.5 Individual raw fluorescence traces by treatment
p_raw_by_treatment <- raw_df %>%
ggplot(aes(x = time_hr, y = value,
group = trace_id, colour = family_id_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ treatment_group) +
scale_colour_manual(values = fam_colours, name = "Family") +
labs(x = "Time (h)", y = "Raw fluorescence (RFU)") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_raw_by_treatment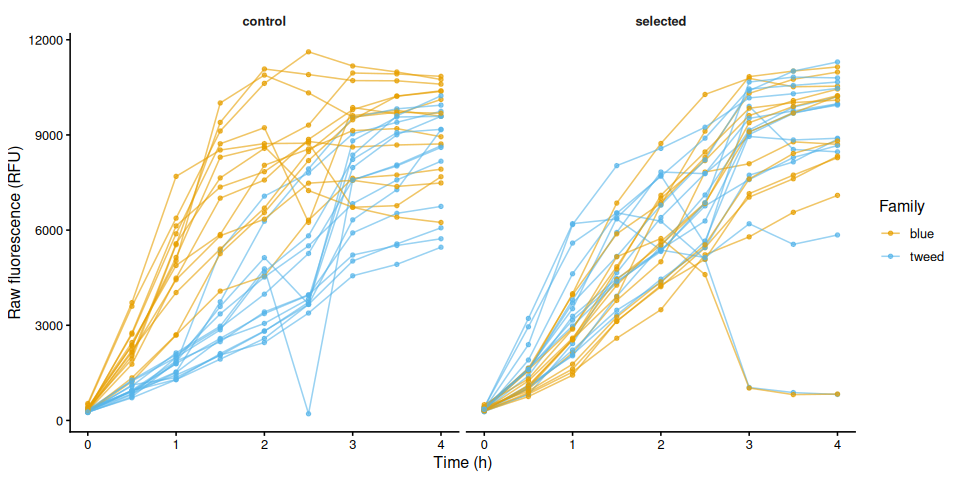
ggsave(file.path(fig_dir, "raw_individual_by_treatment.png"),
p_raw_by_treatment, width = 10, height = 5)6.6 Excluded samples
Wells flagged exclude_from_analysis = TRUE appear in the raw fluorescence plots above but are omitted from all analyses that follow.
excluded_wells <- dat %>%
filter(!is_blank, exclude_from_analysis) %>%
mutate(
sample = if_else(
!is.na(sample_id_group) & trimws(as.character(sample_id_group)) != "",
as.character(sample_id_group),
paste(plate_id, well_id, sep = "_")
)
) %>%
select(plate_id, well_id, sample, family_id_group, treatment_group,
any_of("exclude_reason")) %>%
distinct() %>%
arrange(plate_id, well_id)
if (nrow(excluded_wells) > 0) {
col_names <- c("Plate", "Well", "Sample", "Family", "Treatment")
if ("exclude_reason" %in% names(excluded_wells))
col_names <- c(col_names, "Reason")
cat(knitr::kable(excluded_wells, col.names = col_names), sep = "\n")
} else {
cat("No wells are excluded from analysis.\n")
}| Plate | Well | Sample | Family | Treatment | Reason |
|---|---|---|---|---|---|
| j | B1 | 41 | tweed | selected | volume loss |
| k | A2 | 50 | blue | selected | volume loss |
| l | A4 | 64 | blue | control | volume loss |
7 Blank Correction via Fold-Change Normalization
Following Huffmyer et al.: fluorescence is first expressed as fold-change relative to each well’s own T0 reading (applied to samples and blanks alike), the mean fold-change of blank wells (per plate, per timepoint) is then subtracted. All samples therefore start at exactly 0 at T0 by construction, eliminating the risk of negative starting values from pipetting variance.
7.1 Step 1 – Fold-change relative to T0 for all wells
t0_all <- dat %>%
filter(is.finite(time_hr), is.finite(value)) %>%
group_by(plate_id, well_id) %>%
slice_min(time_hr, n = 1, with_ties = FALSE) %>%
select(plate_id, well_id, value_t0 = value) %>%
ungroup()
dat_fc <- dat %>%
left_join(t0_all, by = c("plate_id", "well_id")) %>%
mutate(fold_change = if_else(
is.finite(value_t0) & value_t0 > 0,
value / value_t0,
NA_real_
))
str(dat_fc)tibble [540 × 18] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:540] "A" "A" "A" "A" ...
$ col_id : int [1:540] 1 2 3 4 1 2 3 4 1 2 ...
$ well_id : chr [1:540] "A1" "A2" "A3" "A4" ...
$ value : num [1:540] 368 350 302 310 316 309 256 336 318 201 ...
$ plate_id : chr [1:540] "h" "h" "h" "h" ...
$ time_hr : num [1:540] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:540] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:540] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_reason : chr [1:540] NA NA NA NA ...
$ family_id_group : chr [1:540] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:540] "13" "14" "15" "16" ...
$ treatment_group : chr [1:540] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:540] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:540] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:540] NA NA NA NA NA NA NA NA NA NA ...
$ area_cm2_measurement : num [1:540] 1.57 1.31 1.1 1.08 1.23 ...
$ value_t0 : num [1:540] 368 350 302 310 316 309 256 336 318 201 ...
$ fold_change : num [1:540] 1 1 1 1 1 1 1 1 1 1 ...7.2 Step 2 – Mean blank fold-change per plate per timepoint
blank_fc_ref <- dat_fc %>%
filter(is_blank) %>%
group_by(plate_id, time_hr) %>%
summarise(mean_blank_fc = mean(fold_change, na.rm = TRUE),
.groups = "drop")
str(blank_fc_ref)tibble [45 × 3] (S3: tbl_df/tbl/data.frame)
$ plate_id : chr [1:45] "h" "h" "h" "h" ...
$ time_hr : num [1:45] 0 0.5 1 1.5 2 2.5 3 3.5 4 0 ...
$ mean_blank_fc: num [1:45] 1 0.99 0.995 0.995 1.01 ...7.3 Step 3 – Subtract blank fold-change from sample fold-change
samples <- dat_fc %>%
filter(!is_blank, !exclude_from_analysis) %>%
mutate(
trace_id = if_else(
!is.na(sample_id_group) & trimws(as.character(sample_id_group)) != "",
as.character(sample_id_group),
paste(plate_id, well_id, sep = "_")
)
) %>%
left_join(blank_fc_ref, by = c("plate_id", "time_hr")) %>%
mutate(corrected_fc = fold_change - mean_blank_fc)
str(samples)tibble [468 × 21] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:468] "A" "A" "A" "A" ...
$ col_id : int [1:468] 1 2 3 4 1 2 3 4 1 3 ...
$ well_id : chr [1:468] "A1" "A2" "A3" "A4" ...
$ value : num [1:468] 368 350 302 310 316 309 256 336 318 317 ...
$ plate_id : chr [1:468] "h" "h" "h" "h" ...
$ time_hr : num [1:468] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:468] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis: logi [1:468] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_reason : chr [1:468] NA NA NA NA ...
$ family_id_group : chr [1:468] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:468] "13" "14" "15" "16" ...
$ treatment_group : chr [1:468] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:468] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement: num [1:468] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement: num [1:468] NA NA NA NA NA NA NA NA NA NA ...
$ area_cm2_measurement : num [1:468] 1.57 1.31 1.1 1.08 1.23 ...
$ value_t0 : num [1:468] 368 350 302 310 316 309 256 336 318 317 ...
$ fold_change : num [1:468] 1 1 1 1 1 1 1 1 1 1 ...
$ trace_id : chr [1:468] "13" "14" "15" "16" ...
$ mean_blank_fc : num [1:468] 1 1 1 1 1 1 1 1 1 1 ...
$ corrected_fc : num [1:468] 0 0 0 0 0 0 0 0 0 0 ...8 Blank-Corrected Fold-Change
8.1 Mean by family
bc_fc_summary <- samples %>%
group_by(family_id_group, treatment_group, time_hr) %>%
summarise(
mean_val = mean(corrected_fc, na.rm = TRUE),
se_val = sd(corrected_fc, na.rm = TRUE) /
sqrt(sum(!is.na(corrected_fc))),
n = sum(!is.na(corrected_fc)),
.groups = "drop"
)
p_bc_fc_mean <- ggplot(bc_fc_summary,
aes(x = time_hr, y = mean_val,
colour = family_id_group, linetype = treatment_group,
group = interaction(family_id_group, treatment_group))) +
geom_ribbon(aes(ymin = mean_val - se_val,
ymax = mean_val + se_val,
fill = family_id_group),
alpha = 0.15, colour = NA) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_colour_manual(values = fam_colours, name = "Family") +
scale_fill_manual(values = fam_colours, name = "Family") +
scale_linetype_manual(values = trt_linetypes, name = "Treatment") +
labs(x = "Time (h)",
y = "Mean blank-corrected fold-change (± SE)") +
theme_classic(base_size = 13)
p_bc_fc_mean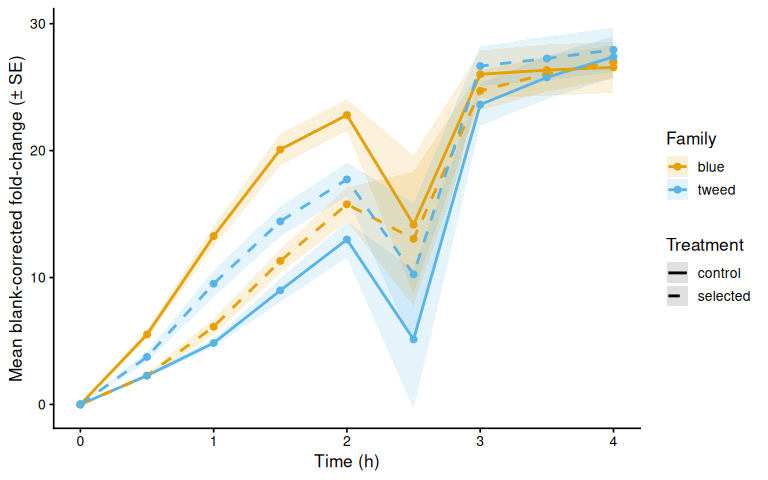
ggsave(file.path(fig_dir, "blank_corrected_fc_mean_by_family.png"),
p_bc_fc_mean, width = 8, height = 5)8.2 Individual traces by family
p_bc_fc_by_family <- samples %>%
ggplot(aes(x = time_hr, y = corrected_fc,
group = trace_id, colour = treatment_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ family_id_group) +
scale_colour_manual(values = trt_colours, name = "Treatment") +
labs(x = "Time (h)", y = "Blank-corrected fold-change") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_bc_fc_by_family
ggsave(file.path(fig_dir, "blank_corrected_fc_by_family.png"),
p_bc_fc_by_family, width = 10, height = 5)8.3 Individual blank-corrected fold-change traces by treatment
p_bc_fc_by_treatment <- samples %>%
ggplot(aes(x = time_hr, y = corrected_fc,
group = trace_id, colour = family_id_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ treatment_group) +
scale_colour_manual(values = fam_colours, name = "Family") +
labs(x = "Time (h)", y = "Blank-corrected fold-change") +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
p_bc_fc_by_treatment
ggsave(file.path(fig_dir, "blank_corrected_fc_by_treatment.png"),
p_bc_fc_by_treatment, width = 10, height = 5)9 Metabolism (Size-Normalised Fold-Change)
Blank-corrected fold-change divided by each active measurement column. This is “metabolism” as defined in Huffmyer et al.
if (length(active_meas_cols) == 0) {
message("No active measurement columns: skipping metabolism calculation.")
metabolism_df <- tibble()
} else {
metabolism_df <- samples
for (mc in active_meas_cols) {
out_col <- paste0("metabolism_per_", mc)
metabolism_df <- metabolism_df %>%
mutate(!!out_col := if_else(
is.finite(.data[[mc]]) & .data[[mc]] > 0 &
is.finite(corrected_fc),
corrected_fc / .data[[mc]],
NA_real_
))
}
}
str(metabolism_df)tibble [468 × 22] (S3: tbl_df/tbl/data.frame)
$ row_id : chr [1:468] "A" "A" "A" "A" ...
$ col_id : int [1:468] 1 2 3 4 1 2 3 4 1 3 ...
$ well_id : chr [1:468] "A1" "A2" "A3" "A4" ...
$ value : num [1:468] 368 350 302 310 316 309 256 336 318 317 ...
$ plate_id : chr [1:468] "h" "h" "h" "h" ...
$ time_hr : num [1:468] 0 0 0 0 0 0 0 0 0 0 ...
$ is_blank : logi [1:468] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_from_analysis : logi [1:468] FALSE FALSE FALSE FALSE FALSE FALSE ...
$ exclude_reason : chr [1:468] NA NA NA NA ...
$ family_id_group : chr [1:468] "tweed" "blue" "tweed" "blue" ...
$ sample_id_group : chr [1:468] "13" "14" "15" "16" ...
$ treatment_group : chr [1:468] "selected" "selected" "control" "control" ...
$ width_mm_measurement : num [1:468] NA NA NA NA NA NA NA NA NA NA ...
$ length_mm_measurement : num [1:468] NA NA NA NA NA NA NA NA NA NA ...
$ weight_mg_measurement : num [1:468] NA NA NA NA NA NA NA NA NA NA ...
$ area_cm2_measurement : num [1:468] 1.57 1.31 1.1 1.08 1.23 ...
$ value_t0 : num [1:468] 368 350 302 310 316 309 256 336 318 317 ...
$ fold_change : num [1:468] 1 1 1 1 1 1 1 1 1 1 ...
$ trace_id : chr [1:468] "13" "14" "15" "16" ...
$ mean_blank_fc : num [1:468] 1 1 1 1 1 1 1 1 1 1 ...
$ corrected_fc : num [1:468] 0 0 0 0 0 0 0 0 0 0 ...
$ metabolism_per_area_cm2_measurement: num [1:468] 0 0 0 0 0 0 0 0 0 0 ...9.1 Mean metabolism by family
if (nrow(metabolism_df) > 0) {
metab_cols <- paste0("metabolism_per_", active_meas_cols)
for (col in metab_cols) {
if (!col %in% names(metabolism_df)) next
mc_label <- str_remove(col, "^metabolism_per_")
metab_summary <- metabolism_df %>%
group_by(family_id_group, treatment_group, time_hr) %>%
summarise(
mean_val = mean(.data[[col]], na.rm = TRUE),
se_val = sd(.data[[col]], na.rm = TRUE) /
sqrt(sum(!is.na(.data[[col]]))),
.groups = "drop"
)
p_metab_mean <- ggplot(metab_summary,
aes(x = time_hr, y = mean_val,
colour = family_id_group, linetype = treatment_group,
group = interaction(family_id_group, treatment_group))) +
geom_ribbon(aes(ymin = mean_val - se_val,
ymax = mean_val + se_val,
fill = family_id_group),
alpha = 0.15, colour = NA) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_colour_manual(values = fam_colours, name = "Family") +
scale_fill_manual(values = fam_colours, name = "Family") +
scale_linetype_manual(values = trt_linetypes, name = "Treatment") +
labs(x = "Time (h)",
y = paste0(metabolism_y_label(col), " (± SE)")) +
theme_classic(base_size = 13)
print(p_metab_mean)
ggsave(
file.path(fig_dir,
paste0("metabolism_mean_", mc_label, ".png")),
p_metab_mean, width = 8, height = 5)
}
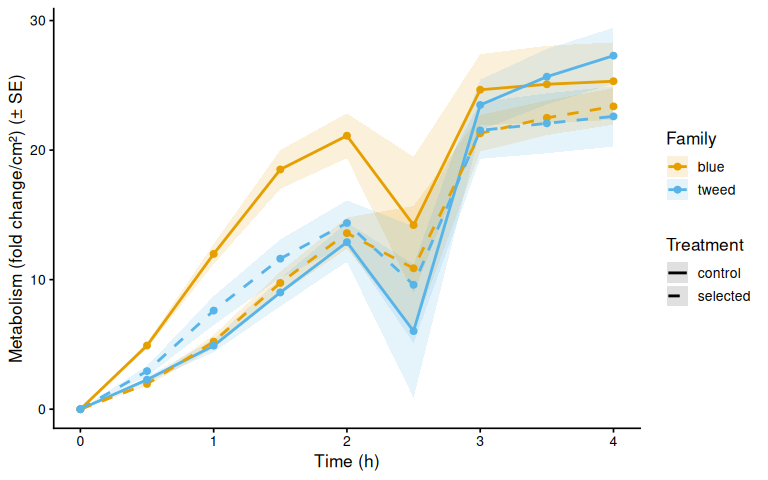
}
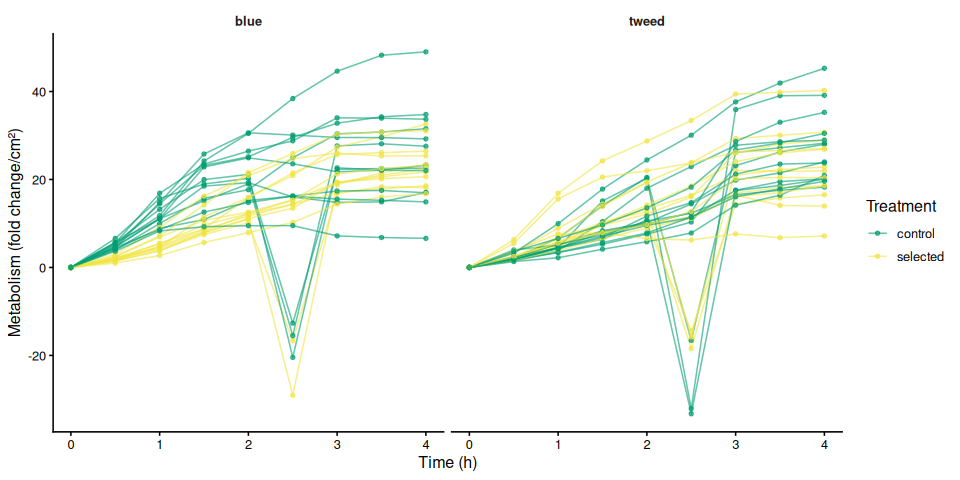
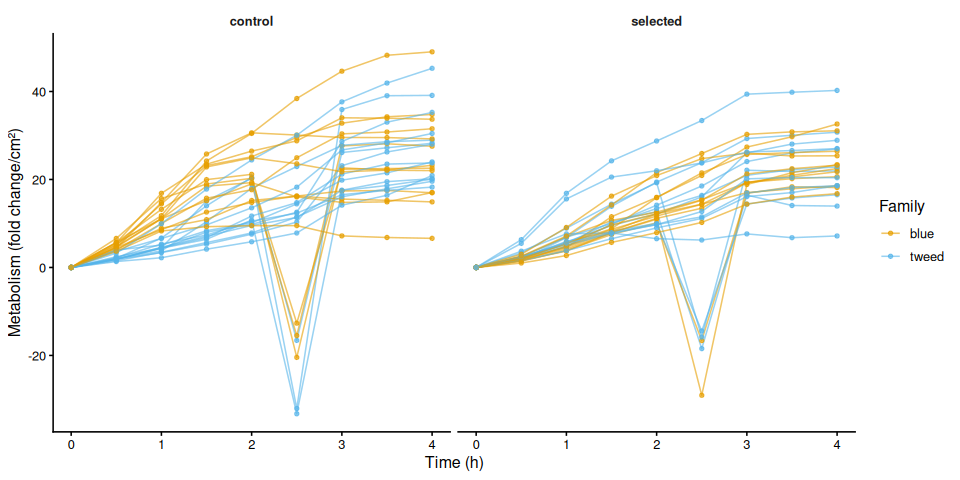
9.2 Individual metabolism traces by family
if (nrow(metabolism_df) > 0) {
for (col in metab_cols) {
if (!col %in% names(metabolism_df)) next
mc_label <- str_remove(col, "^metabolism_per_")
p_metab_by_family <- ggplot(metabolism_df,
aes(x = time_hr, y = .data[[col]],
group = trace_id, colour = treatment_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ family_id_group) +
scale_colour_manual(values = trt_colours, name = "Treatment") +
labs(x = "Time (h)", y = metabolism_y_label(col)) +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
print(p_metab_by_family)
ggsave(
file.path(fig_dir,
paste0("metabolism_individual_", mc_label, "_by_family.png")),
p_metab_by_family, width = 10, height = 5)
p_metab_by_treatment <- ggplot(metabolism_df,
aes(x = time_hr, y = .data[[col]],
group = trace_id, colour = family_id_group)) +
geom_line(alpha = 0.6) +
geom_point(size = 1.2, alpha = 0.7) +
facet_wrap(~ treatment_group) +
scale_colour_manual(values = fam_colours, name = "Family") +
labs(x = "Time (h)", y = metabolism_y_label(col)) +
theme_classic(base_size = 12) +
theme(strip.background = element_blank(),
strip.text = element_text(face = "bold"))
print(p_metab_by_treatment)
ggsave(
file.path(fig_dir,
paste0("metabolism_individual_", mc_label, "_by_treatment.png")),
p_metab_by_treatment, width = 10, height = 5)
}
}
10 Time-Series Statistical Analysis
Linear mixed effects models test the effect of experimental variables on metabolism over time. Individual (sample_id_group) is included as a random intercept to account for repeated measures across timepoints. Type III ANOVA with Satterthwaite’s approximation (lmerTest) assesses significance; post-hoc pairwise comparisons use estimated marginal means (emmeans, Tukey adjustment).
run_ts_stats <- function(df, value_col) {
has_family <- "family_id_group" %in% names(df) &&
length(unique(na.omit(df$family_id_group))) > 1
has_treatment <- "treatment_group" %in% names(df) &&
length(unique(na.omit(df$treatment_group))) > 1
if (!has_family && !has_treatment) return(NULL)
df <- df %>%
filter(is.finite(.data[[value_col]]), is.finite(time_hr)) %>%
mutate(
time_f = factor(time_hr),
individual = factor(trace_id)
)
if (nrow(df) == 0) return(NULL)
if (has_family) df <- df %>% mutate(family = factor(family_id_group))
if (has_treatment) df <- df %>% mutate(treatment = factor(treatment_group))
if (has_family && length(unique(na.omit(df$family))) < 2) return(NULL)
if (has_treatment && length(unique(na.omit(df$treatment))) < 2) return(NULL)
fixed <- if (has_family && has_treatment)
paste0(value_col, " ~ time_f * family * treatment")
else if (has_family)
paste0(value_col, " ~ time_f * family")
else
paste0(value_col, " ~ time_f * treatment")
model <- lmer(
as.formula(paste0(fixed, " + (1 | individual)")),
data = df
)
anova_res <- anova(model, type = 3, ddf = "Satterthwaite")
# Pairwise comparisons of group combinations at each timepoint
emm_spec <- if (has_family && has_treatment)
~ family * treatment | time_f
else if (has_family)
~ family | time_f
else
~ treatment | time_f
emm <- emmeans(model, emm_spec)
pairs_res <- as.data.frame(pairs(emm, adjust = "tukey"))
# Main-effect marginal means (collapsed across time)
emm_main <- if (has_family && has_treatment)
emmeans(model, ~ family * treatment)
else if (has_family)
emmeans(model, ~ family)
else
emmeans(model, ~ treatment)
pairs_main <- as.data.frame(pairs(emm_main, adjust = "tukey"))
list(
model = model,
anova = anova_res,
pairs_by_time = pairs_res,
pairs_main = pairs_main,
has_family = has_family,
has_treatment = has_treatment
)
}
ts_stats <- list()
if (nrow(metabolism_df) > 0) {
for (mc in active_meas_cols) {
col <- paste0("metabolism_per_", mc)
if (col %in% names(metabolism_df))
ts_stats[[col]] <- run_ts_stats(metabolism_df, col)
}
}10.1 Results
for (col in names(ts_stats)) {
res <- ts_stats[[col]]
if (is.null(res)) next
cat("\n\n----\n### Metric:", col, "\n\n")
cat("**Type III ANOVA (Satterthwaite approximation):**\n")
print(res$anova)
cat("\n**Marginal means – main effects (collapsed across time):**\n")
print(res$pairs_main)
cat("\n**Pairwise comparisons by timepoint (Tukey):**\n")
print(res$pairs_by_time)
}Metric: metabolism_per_area_cm2_measurement
Signif. codes: 0 ‘’ 0.001 ’’ 0.01 ’’ 0.05 ‘.’ 0.1 ’ ’ 1
Marginal means – main effects (collapsed across time): contrast estimate SE df t.ratio p.value blue control - tweed control 3.803467 1.925512 48 1.975 0.2116 blue control - blue selected 4.137359 2.001279 48 2.067 0.1785 blue control - tweed selected 3.716759 1.960845 48 1.895 0.2436 tweed control - blue selected 0.333892 1.966672 48 0.170 0.9982 tweed control - tweed selected -0.086707 1.925512 48 -0.045 1.0000 blue selected - tweed selected -0.420600 2.001279 48 -0.210 0.9967
Results are averaged over the levels of: time_f Degrees-of-freedom method: kenward-roger P value adjustment: tukey method for comparing a family of 4 estimates
Pairwise comparisons by timepoint (Tukey): time_f = 0: contrast estimate SE df t.ratio p.value blue control - tweed control 0.000000 3.093522 243.65 0.000 1.0000 blue control - blue selected 0.000000 3.215250 243.65 0.000 1.0000 blue control - tweed selected 0.000000 3.150289 243.65 0.000 1.0000 tweed control - blue selected 0.000000 3.159651 243.65 0.000 1.0000 tweed control - tweed selected 0.000000 3.093522 243.65 0.000 1.0000 blue selected - tweed selected 0.000000 3.215250 243.65 0.000 1.0000
time_f = 0.5: contrast estimate SE df t.ratio p.value blue control - tweed control 2.637174 3.093522 243.65 0.852 0.8292 blue control - blue selected 2.966957 3.215250 243.65 0.923 0.7927 blue control - tweed selected 1.975025 3.150289 243.65 0.627 0.9233 tweed control - blue selected 0.329783 3.159651 243.65 0.104 0.9996 tweed control - tweed selected -0.662149 3.093522 243.65 -0.214 0.9965 blue selected - tweed selected -0.991933 3.215250 243.65 -0.309 0.9898
time_f = 1: contrast estimate SE df t.ratio p.value blue control - tweed control 7.081608 3.093522 243.65 2.289 0.1034 blue control - blue selected 6.768109 3.215250 243.65 2.105 0.1544 blue control - tweed selected 4.373666 3.150289 243.65 1.388 0.5079 tweed control - blue selected -0.313499 3.159651 243.65 -0.099 0.9996 tweed control - tweed selected -2.707942 3.093522 243.65 -0.875 0.8176 blue selected - tweed selected -2.394443 3.215250 243.65 -0.745 0.8788
time_f = 1.5: contrast estimate SE df t.ratio p.value blue control - tweed control 9.493672 3.093522 243.65 3.069 0.0127 blue control - blue selected 8.755837 3.215250 243.65 2.723 0.0348 blue control - tweed selected 6.885311 3.150289 243.65 2.186 0.1301 tweed control - blue selected -0.737835 3.159651 243.65 -0.234 0.9955 tweed control - tweed selected -2.608361 3.093522 243.65 -0.843 0.8338 blue selected - tweed selected -1.870526 3.215250 243.65 -0.582 0.9375
time_f = 2: contrast estimate SE df t.ratio p.value blue control - tweed control 8.217219 3.093522 243.65 2.656 0.0416 blue control - blue selected 7.519209 3.215250 243.65 2.339 0.0922 blue control - tweed selected 6.739129 3.150289 243.65 2.139 0.1437 tweed control - blue selected -0.698011 3.159651 243.65 -0.221 0.9962 tweed control - tweed selected -1.478090 3.093522 243.65 -0.478 0.9639 blue selected - tweed selected -0.780079 3.215250 243.65 -0.243 0.9950
time_f = 2.5: contrast estimate SE df t.ratio p.value blue control - tweed control 8.182513 3.093522 243.65 2.645 0.0429 blue control - blue selected 3.334347 3.215250 243.65 1.037 0.7279 blue control - tweed selected 4.610040 3.150289 243.65 1.463 0.4613 tweed control - blue selected -4.848166 3.159651 243.65 -1.534 0.4185 tweed control - tweed selected -3.572473 3.093522 243.65 -1.155 0.6559 blue selected - tweed selected 1.275693 3.215250 243.65 0.397 0.9788
time_f = 3: contrast estimate SE df t.ratio p.value blue control - tweed control 1.173536 3.093522 243.65 0.379 0.9814 blue control - blue selected 3.358980 3.215250 243.65 1.045 0.7233 blue control - tweed selected 3.136225 3.150289 243.65 0.996 0.7521 tweed control - blue selected 2.185444 3.159651 243.65 0.692 0.9002 tweed control - tweed selected 1.962689 3.093522 243.65 0.634 0.9208 blue selected - tweed selected -0.222756 3.215250 243.65 -0.069 0.9999
time_f = 3.5: contrast estimate SE df t.ratio p.value blue control - tweed control -0.583554 3.093522 243.65 -0.189 0.9976 blue control - blue selected 2.588310 3.215250 243.65 0.805 0.8520 blue control - tweed selected 3.016235 3.150289 243.65 0.957 0.7737 tweed control - blue selected 3.171863 3.159651 243.65 1.004 0.7473 tweed control - tweed selected 3.599788 3.093522 243.65 1.164 0.6503 blue selected - tweed selected 0.427925 3.215250 243.65 0.133 0.9992
time_f = 4: contrast estimate SE df t.ratio p.value blue control - tweed control -1.970967 3.093522 243.65 -0.637 0.9199 blue control - blue selected 1.944482 3.215250 243.65 0.605 0.9305 blue control - tweed selected 2.715204 3.150289 243.65 0.862 0.8245 tweed control - blue selected 3.915449 3.159651 243.65 1.239 0.6025 tweed control - tweed selected 4.686171 3.093522 243.65 1.515 0.4302 blue selected - tweed selected 0.770722 3.215250 243.65 0.240 0.9951
Degrees-of-freedom method: kenward-roger P value adjustment: tukey method for comparing a family of 4 estimates
Area Under the Curve (AUC)
AUC computed per individual via the trapezoid rule across all timepoints. metabolism_per_* is the primary metric matching the paper; corrected_fc and raw_fluorescence are retained for reference.
compute_auc <- function(df, value_col, group_vars) { df %>% filter(is.finite(time_hr), is.finite(.data[[value_col]])) %>% group_by(across(all_of(group_vars))) %>% summarise( AUC = trapezoid_auc(time_hr, .data[[value_col]]), n_timepoints = n(), .groups = “drop” ) %>% filter(is.finite(AUC)) }
\# Only include grouping columns that are actually present in the data individual_vars <- intersect( c(“trace_id”, “family_id_group”, “treatment_group”), names(metabolism_df) )
auc_metab_list <- list() if (nrow(metabolism_df) > 0) { for (mc in active_meas_cols) { col <- paste0(“metabolism_per_”, mc) if (col %in% names(metabolism_df)) { auc_metab_list[[col]] <- compute_auc(metabolism_df, col, individual_vars) %>% mutate(metric = col) } } }
auc_all <- bind_rows(auc_metab_list)
str(auc_all)tibble [52 × 6] (S3: tbl_df/tbl/data.frame) $ trace_id : chr [1:52] "13" "14" "15" "16" ... $ family_id_group: chr [1:52] "tweed" "blue" "tweed" "blue" ... $ treatment_group: chr [1:52] "selected" "selected" "control" "control" ... $ AUC : num [1:52] 24.9 25.3 46.3 52.5 42.5 ... $ n_timepoints : int [1:52] 9 9 9 9 9 9 9 9 9 9 ... $ metric : chr [1:52] "metabolism_per_area_cm2_measurement" "metabolism_per_area_cm2_measurement" "metabolism_per_area_cm2_measurement" "metabolism_per_area_cm2_measurement" ...AUC summary tables
sum_vars <- intersect( c(“metric”, “family_id_group”, “treatment_group”), names(auc_all) ) auc_summary <- auc_all %>% group_by(across(all_of(sum_vars))) %>% summarise( n = n(), mean = mean(AUC, na.rm = TRUE), sd = sd(AUC, na.rm = TRUE), se = sd / sqrt(n), median = median(AUC, na.rm = TRUE), .groups = “drop” )
print(auc_summary)# A tibble: 4 × 8 metric family_id_group treatment_group n mean sd se median <chr> <chr> <chr> <int> <dbl> <dbl> <dbl> <dbl> 1 metabolism_per… blue control 13 66.6 24.4 6.76 53.8 2 metabolism_per… blue selected 12 48.4 16.1 4.65 47.4 3 metabolism_per… tweed control 14 49.0 17.6 4.72 47.0 4 metabolism_per… tweed selected 13 50.5 23.1 6.40 44.6Statistical Analysis
Each individual clam (sample_id_group) is the observational unit. The model is built from whichever grouping factors are present: both family and treatment (with interaction) when both exist, or a one-way model when only one factor is available. Each plate maps to a unique family × treatment combination, so plate-level and group-level variance are confounded; interpret accordingly.
has_treatment <- “treatment_group” %in% names(auc_df) && length(unique(na.omit(auc_df\(treatment_group))) > 1 if (!has_family && !has_treatment) { return(list(model = NULL, anova = NULL, pairs_full = empty, pairs_family = empty, pairs_trt = empty, has_family = FALSE, has_treatment = FALSE)) } if (has_family) auc_df <- auc_df %>% mutate(family = factor(family_id_group)) if (has_treatment) auc_df <- auc_df %>% mutate(treatment = factor(treatment_group)) formula_str <- if (has_family && has_treatment) “AUC ~ family * treatment” else if (has_family) “AUC ~ family” else “AUC ~ treatment” model <- lm(as.formula(formula_str), data = auc_df) anova_res <- anova(model) if (has_family && has_treatment) { pairs_full <- as.data.frame(pairs(emmeans(model, ~ family * treatment), adjust = “tukey”)) pairs_family <- as.data.frame(pairs(emmeans(model, ~ family), adjust = “tukey”)) pairs_trt <- as.data.frame(pairs(emmeans(model, ~ treatment), adjust = “tukey”)) } else if (has_family) { pairs_family <- as.data.frame(pairs(emmeans(model, ~ family), adjust = “tukey”)) pairs_full <- pairs_family pairs_trt <- empty } else { pairs_trt <- as.data.frame(pairs(emmeans(model, ~ treatment), adjust = “tukey”)) pairs_full <- pairs_trt pairs_family <- empty } list( model = model, anova = anova_res, pairs_full = pairs_full, pairs_family = pairs_family, pairs_trt = pairs_trt, has_family = has_family, has_treatment = has_treatment ) } metrics_to_test <- unique(auc_all\)metric) stats_results <- map( set_names(metrics_to_test), ~ run_auc_stats(auc_all %>% filter(metric == .x)) ) ``` ## Results by metric
10.1.1 Metric: metabolism_per_area_cm2_measurement
ANOVA: Analysis of Variance Table
Response: AUC Df Sum Sq Mean Sq F value Pr(>F)
family 1 862.9 862.86 2.0268 0.16101
treatment 1 811.2 811.21 1.9055 0.17386
family:treatment 1 1256.7 1256.75 2.9520 0.09222 . Residuals 48 20434.7 425.72
— Signif. codes: 0 ‘’ 0.001 ’’ 0.01 ’’ 0.05 ‘.’ 0.1 ’ ’ 1
Pairwise: family × treatment (Tukey): contrast estimate SE df t.ratio p.value blue control - tweed control 17.608342 7.947125 48 2.216 0.1336 blue control - blue selected 18.131995 8.259839 48 2.195 0.1392 blue control - tweed selected 16.046616 8.092956 48 1.983 0.2087 tweed control - blue selected 0.523652 8.117007 48 0.065 0.9999 tweed control - tweed selected -1.561726 7.947125 48 -0.197 0.9973 blue selected - tweed selected -2.085379 8.259839 48 -0.252 0.9943
P value adjustment: tukey method for comparing a family of 4 estimates
Pairwise: family main effect: contrast estimate SE df t.ratio p.value blue - tweed 7.761482 5.731094 48 1.354 0.1820
Results are averaged over the levels of: treatment
Pairwise: treatment main effect: contrast estimate SE df t.ratio p.value control - selected 8.285134 5.731094 48 1.446 0.1548
Results are averaged over the levels of: family
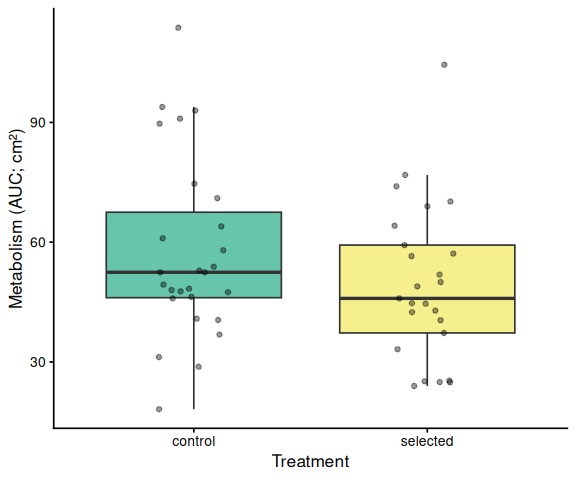
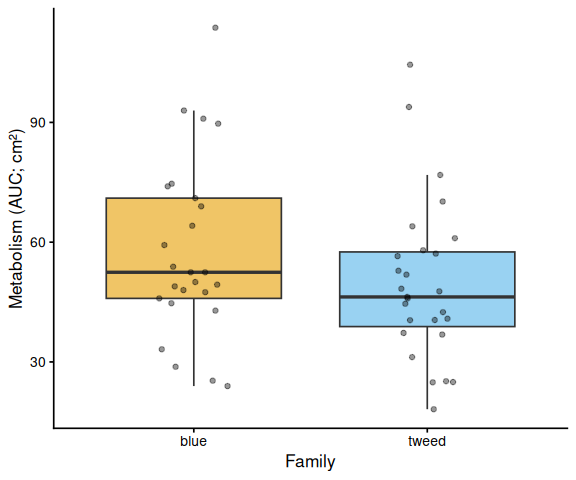
11 AUC Box Plots with Statistical Annotations
Significance labels: *** p < 0.001, ** p < 0.01, * p < 0.05. Brackets are drawn only for significant pairs (p < 0.05). Plots are generated for whichever grouping factors are present: treatment-only, family-only, all-groups, within-family, and within-treatment.
sig_label <- function(p) {
case_when(p < 0.001 ~ "***", p < 0.01 ~ "**", p < 0.05 ~ "*",
TRUE ~ "ns")
}
# Add significance brackets to an existing ggplot.
# pairs_df : data frame with $contrast and $p.value columns
# group_levels: ordered character vector matching x-axis factor levels
# y_vals : numeric vector of AUC values used to set bracket heights
add_sig_brackets <- function(p, pairs_df, group_levels, y_vals) {
sig_pairs <- pairs_df %>%
mutate(label = sig_label(p.value)) %>%
filter(label != "ns")
if (nrow(sig_pairs) == 0) return(p)
y_max <- max(y_vals, na.rm = TRUE)
y_range <- diff(range(y_vals, na.rm = TRUE))
step <- y_range * 0.12
for (i in seq_len(nrow(sig_pairs))) {
parts <- str_split(as.character(sig_pairs$contrast[i]), " - ", 2)[[1]]
g1 <- trimws(parts[1])
g2 <- trimws(parts[2])
x1 <- match(g1, group_levels)
x2 <- match(g2, group_levels)
if (is.na(x1) || is.na(x2)) next
bar_y <- y_max + i * step
p <- p +
annotate("segment", x = x1, xend = x2,
y = bar_y, yend = bar_y,
colour = "black", linewidth = 0.6) +
annotate("segment", x = x1, xend = x1,
y = bar_y, yend = bar_y - step * 0.3,
colour = "black", linewidth = 0.6) +
annotate("segment", x = x2, xend = x2,
y = bar_y, yend = bar_y - step * 0.3,
colour = "black", linewidth = 0.6) +
annotate("text", x = (x1 + x2) / 2,
y = bar_y + step * 0.15,
label = sig_pairs$label[i], size = 4.5)
}
p
}for (met in metrics_to_test) {
df <- auc_all %>% filter(metric == met)
stats <- stats_results[[met]]
y_lab <- auc_y_label(met)
has_fam <- stats$has_family
has_trt <- stats$has_treatment
# ── Treatment main effect (x = treatment, tick = treatment name) ───────
if (has_trt) {
df_p <- df %>%
mutate(x = factor(treatment_group, levels = sort(unique(treatment_group))))
grps <- levels(df_p$x)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = trt_colours[grps], guide = "none") +
labs(x = "Treatment", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, stats$pairs_trt, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_treatment_", met, ".png")),
p, width = 5, height = 5)
}
# ── Family main effect (x = family, tick = family name) ───────────────
if (has_fam) {
df_p <- df %>%
mutate(x = factor(family_id_group, levels = sort(unique(family_id_group))))
grps <- levels(df_p$x)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fam_colours[grps], guide = "none") +
labs(x = "Family", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, stats$pairs_family, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_family_", met, ".png")),
p, width = 5, height = 5)
}
# Remaining plots require both factors
if (!has_fam || !has_trt) next
# ── All family:treatment groups (x = family:treatment) ─────────────────
# emmeans contrasts use spaces; convert to colon to match tick labels
pairs_fc <- stats$pairs_full %>%
mutate(contrast = str_replace_all(
contrast,
"([a-z]+) ([a-z]+)( - )([a-z]+) ([a-z]+)",
"\\1:\\2\\3\\4:\\5"
))
df_p <- df %>%
mutate(x = factor(
paste(family_id_group, treatment_group, sep = ":"),
levels = sort(unique(paste(family_id_group, treatment_group, sep = ":")))
))
grps <- levels(df_p$x)
fill_map <- setNames(make_palette(length(grps)), grps)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fill_map, guide = "none") +
labs(x = "Family : Treatment", y = y_lab) +
theme_classic(base_size = 13) +
theme(axis.text.x = element_text(angle = 20, hjust = 1))
p <- add_sig_brackets(p, pairs_fc, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_all_groups_", met, ".png")),
p, width = 6, height = 5)
# ── Within each family: treatment comparison (x = family:treatment) ────
# Tick labels are family:treatment so these plots are visually distinct
# from the treatment main-effect plot above.
for (fam in sort(unique(df$family_id_group))) {
df_p <- df %>%
filter(family_id_group == fam) %>%
mutate(x = factor(
paste(family_id_group, treatment_group, sep = ":"),
levels = sort(unique(paste(family_id_group, treatment_group, sep = ":")))
))
grps <- levels(df_p$x)
pairs_sub <- pairs_fc %>%
filter(str_count(contrast, paste0(fam, ":")) == 2)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fill_map[grps], guide = "none") +
labs(x = "Family : Treatment", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, pairs_sub, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_", fam, "_trt_", met, ".png")),
p, width = 5, height = 5)
}
# ── Within each treatment: family comparison (x = family:treatment) ────
# Tick labels are family:treatment so these plots are visually distinct
# from the family main-effect plot above.
for (trt in sort(unique(df$treatment_group))) {
df_p <- df %>%
filter(treatment_group == trt) %>%
mutate(x = factor(
paste(family_id_group, treatment_group, sep = ":"),
levels = sort(unique(paste(family_id_group, treatment_group, sep = ":")))
))
grps <- levels(df_p$x)
pairs_sub <- pairs_fc %>%
filter(str_count(contrast, paste0(":", trt)) == 2)
p <- ggplot(df_p, aes(x = x, y = AUC, fill = x)) +
geom_boxplot(alpha = 0.6, outlier.shape = NA) +
geom_jitter(width = 0.15, alpha = 0.4, size = 1.5) +
scale_fill_manual(values = fill_map[grps], guide = "none") +
labs(x = "Family : Treatment", y = y_lab) +
theme_classic(base_size = 13)
p <- add_sig_brackets(p, pairs_sub, grps, df_p$AUC)
print(p)
ggsave(file.path(fig_dir, paste0("auc_", trt, "_fam_", met, ".png")),
p, width = 5, height = 5)
}
}
12 Save Output Data
write_csv(auc_all, file.path(out_dir, "auc_all_metrics.csv"))
write_csv(auc_summary, file.path(out_dir, "auc_summary.csv"))
if (nrow(metabolism_df) > 0)
write_csv(metabolism_df,
file.path(out_dir, "metabolism.csv"))
stats_compiled <- map_dfr(metrics_to_test, function(met) {
bind_rows(
stats_results[[met]]$pairs_full %>%
mutate(comparison = "family:treatment"),
stats_results[[met]]$pairs_family %>%
mutate(comparison = "family"),
stats_results[[met]]$pairs_trt %>%
mutate(comparison = "treatment")
) %>% mutate(metric = met)
})
write_csv(stats_compiled,
file.path(out_dir, "pairwise_stats.csv"))
message("Output files written to: ", out_dir)MATERIALS & METHODS
Clams were processed in 12-well plates and submerged in 5.0mL of resazurin working solution stored at 4oC,prepared on 20260330 (notebook entry).
Plates were measured every 30mins on a Synergy HTX (Agilent) plate reader.
Plate layout was randomized; Steven is currently the only person who knows the treatment assignments.
The following wells lost volume - possibly due to clam spitting?
- GA3
- JB1
- KA2
- LA4
Plate Temps
Wells were spot checked and range noted in table below.
| PLATE | TIME | TEMP |
|---|---|---|
| G | 0 | 11-12 |
| G | 0.5 | 14-16 |
| G | 1 | 24-30 |
| G | 1.5 | 27-30 |
| G | 2 | 31-32 |
| G | 2.5 | 31-32 |
| G | 3 | 31-32 |
| G | 3.5 | 31-32 |
| G | 4 | 33 |
RESULTS
Plate G was automatically excluded from all analyses: its T0 read was accidentally exported as a 24-well plate, causing it to fail the consistency check. Three additional wells were excluded due to volume loss: J B1 (sample 41, tweed selected), K A2 (sample 50, blue selected), and L A4 (sample 64, blue control).
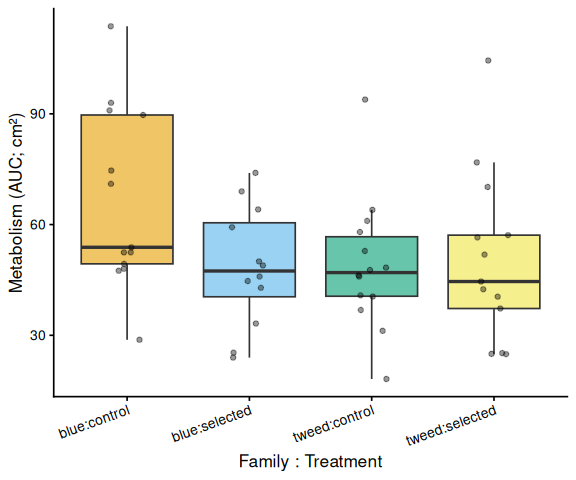
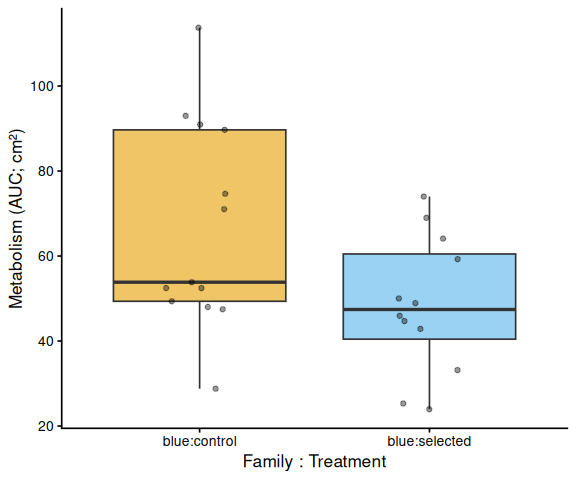
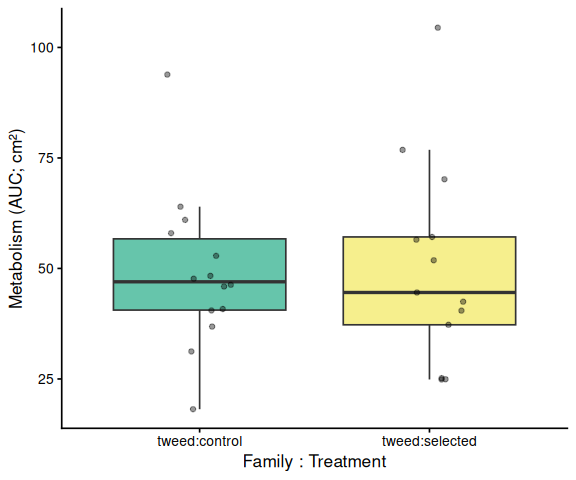
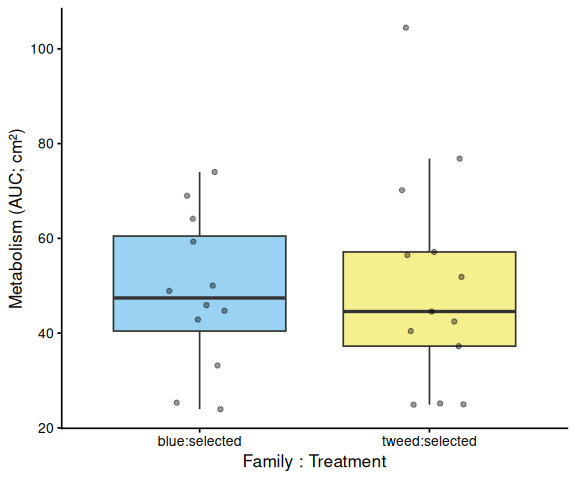
Metabolic activity (AUC of blank-corrected, size-normalised fold-change per cm² shell area) did not differ significantly by family, treatment, or their interaction (ANOVA: family F = 2.03, p = 0.161; treatment F = 1.91, p = 0.174; family × treatment F = 2.95, p = 0.092). Blue control clams had the highest mean AUC (66.6 ± 6.8 SE), while the other three groups were similar: tweed control (49.0 ± 4.7), tweed selected (50.5 ± 6.4), and blue selected (48.4 ± 4.7). No pairwise contrast reached significance after Tukey adjustment (all p > 0.13).
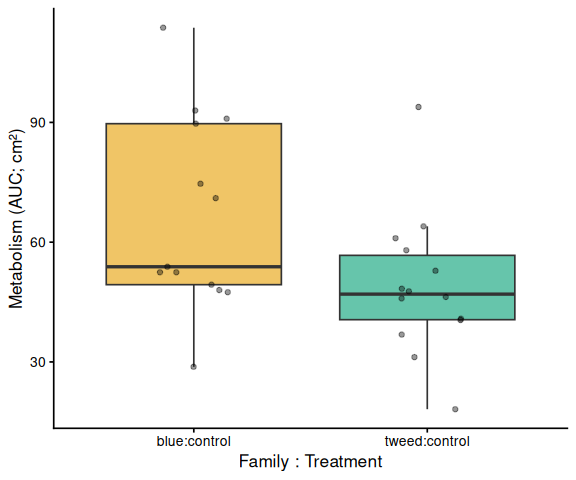
The time-series mixed-effects model identified a transient divergence of blue control clams at mid-assay timepoints:
- t = 1.5 h: blue control > tweed control (p = 0.013) and > blue selected (p = 0.035)
- t = 2.0 h: blue control > tweed control (p = 0.042)
- t = 2.5 h: blue control > tweed control (p = 0.043)
No significant differences were detected at any other timepoints (all Tukey-adjusted p > 0.09).
In summary, selected clams did not show higher metabolic activity than control clams in this larger-clam 36°C assay. The primary signal in this experiment is a transient elevation in blue-family control clams at 1.5–2.5 h, not a treatment effect; the marginally non-significant family × treatment interaction (p = 0.092) is consistent with this blue-control-specific pattern. This same blue-control divergence was also observed on Day 0 of the smaller-clam 36°C experiment.