— UPDATE 20171009 —
Having run through this a bunch of times now, I realized that the analysis below incorrectly identifies the outputs from Sean’s Redundans runs. The correct output from each of those runs should be the “scaffolds.reduced.fa” FAST files. The “contigs.fa” files that I linked to below are actually the assemblies produced by other programs; which are required as an input for Redudans.
I recently completed an assembly of the UW PacBio sequencing data using Racon and wanted some assembly stats, as well as a way to compare this assembly to the assemblies Sean had completed.
Additionally, Steven recently performed an assembly comparison and I noticed he got some odd results. Specifically, of the three assemblies he compared (PacBio x 1, Illumina x 2), both of the Illumina assemblies had a large quantity of “Ns” in the assemblies. This didn’t seem right and the comparison program he used (QUAST) spit out a message indicating that it seemed like scaffolds were used, instead of contigs. So, I thought I’d give it a shot and see if I could track down non-scaffolded assemblies produced by Sean.
Jupyter notebook (GitHub): 20171003_docker_oly_assembly_comparisons.ipynb
First, I compared the following six assemblies (FASTA files) using QUAST:
Sean’s Assemblies:
- PacBio (Canu): http://owl.fish.washington.edu/scaphapoda/Sean/Oly_Canu_Output/oly_pacbio_.contigs.fasta
- Illumina (Platanus): http://owl.fish.washington.edu/scaphapoda/Sean/Oly_Illumina_Platanus_Assembly/Oly_Out__contig.fa
- Illumina (Platanus): http://owl.fish.washington.edu/scaphapoda/Sean/Oly_Platanus_Assembly_Kmer-22/Oly_Out__contig.fa
- Illumina/PacBio (Redundans): http://owl.fish.washington.edu/scaphapoda/Sean/Oly_Redundans_Output/contigs.fa
- Illumina/PacBio (Redundans): http://owl.fish.washington.edu/scaphapoda/Sean/Oly_Redundans_Output_Try_2/contigs.fa
Sam’s Assembly:
- PacBio (Racon): http://owl.fish.washington.edu/Athaliana/201709_oly_pacbio_assembly_minimap_asm_racon/20170918_oly_pacbio_racon1_consensus.fasta
QUAST output directory: http://owl.fish.washington.edu/Athaliana/20171003_quast_oly_genome_assemblies/
Here’s the assembly comparison of all assemblies (click on image for larger view):
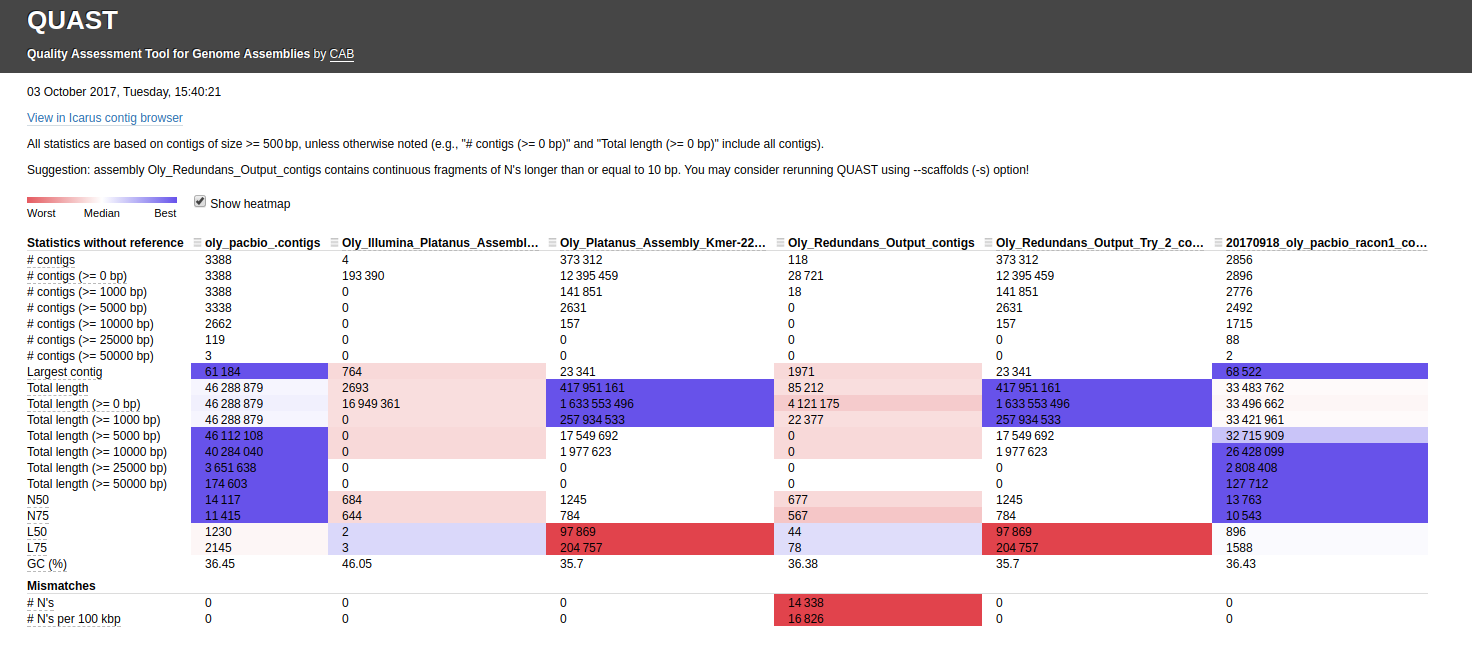
Interactive version of that graphic is here: http://owl.fish.washington.edu/Athaliana/20171003_quast_oly_genome_assemblies/report.html
The first thing that jumps out to me is the fact that two of the Illumina assemblies, which used different assemblers(!!) have the EXACT same assembly stats. This occurrence seems extremely unlikely. I’ve double-checked my Jupyter notebook to make sure that I didn’t assign the same file by accident (see Input #6)

Very strange!
I also noticed that the first Redundans assembly of Sean’s has a ton of “Ns”, suggesting that it’s actually a scaffolded assembly. As with Steven’s QUAST run, QUAST spits out the messages suggesting to use the “–scaffold” option for this file.
The other thing I noticed is the two PacBio assemblies (Canu & Racon) have a huge difference in the total number of bp (~13,000,000)! I ran a QUAST assembly comparison between just those two for easier viewing/comparison (http://owl.fish.washington.edu/Athaliana/20171003_quast_oly_pacbio_assemblies/):
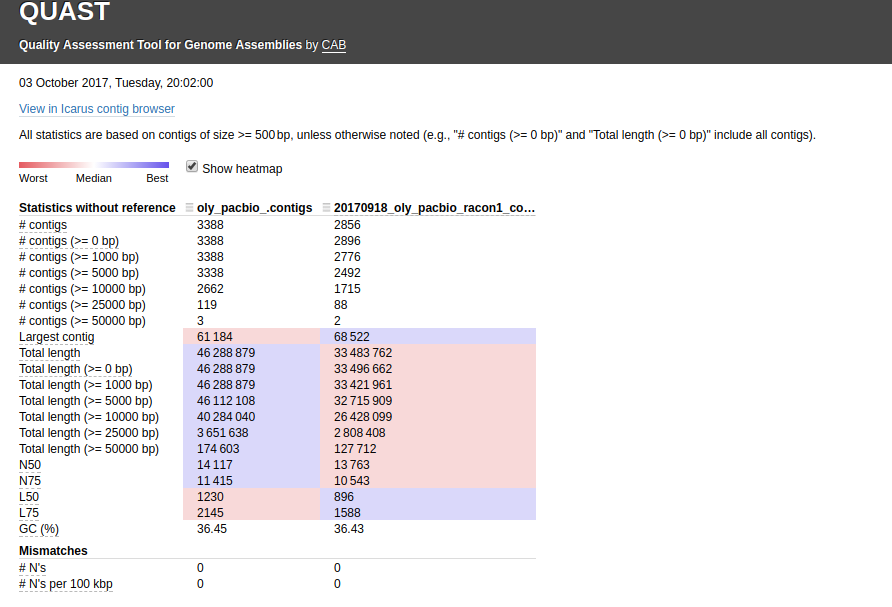
Interactive version of that graphic is here: http://owl.fish.washington.edu/Athaliana/20171003_quast_oly_pacbio_assemblies/report.html
The fact that there is such a large discrepancy in the total number of bps between these two assemblies really leaves me to believe that I am missing a FASTQ file from my assembly. I’m going to go back and see if that is indeed the case or if this difference in the assemblies is real.
Here’s an embedded version of my Jupyter notebook:







