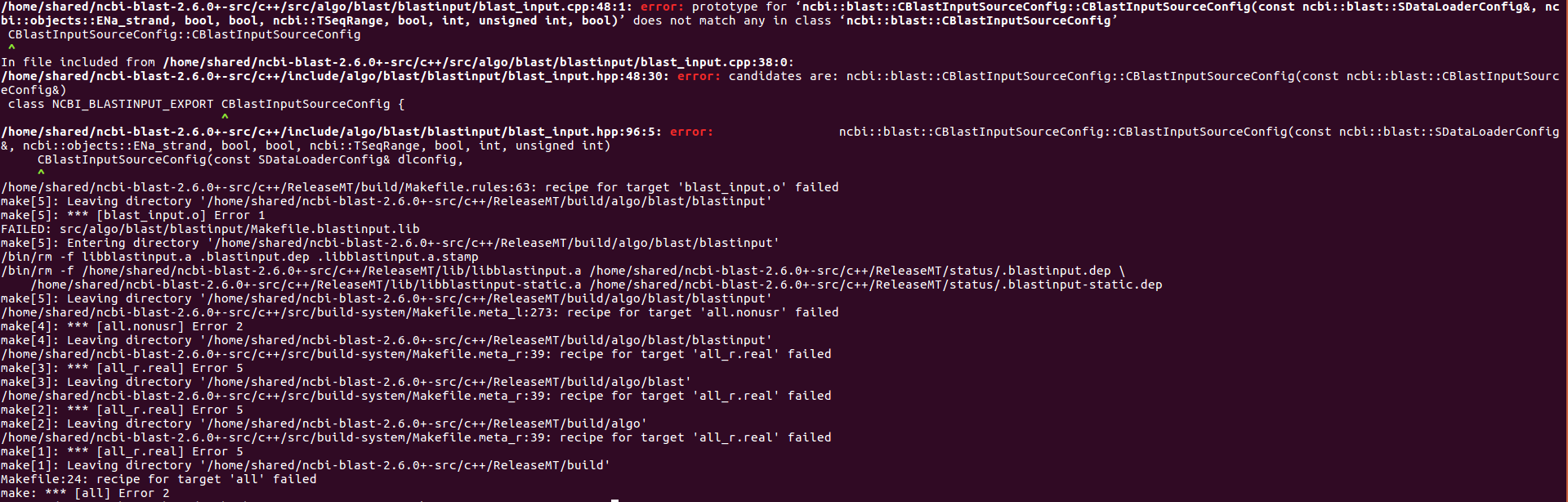
After yesterday’s difficulties getting RMblast to compile, I deleted the folder and went through the build process again.
This time it worked, but it did not put rmblastn in the specified location (/home/shared/rmblast).
This fact took me a fair amount of time to figure out. Finally, after a couple of different re-builds, I ran find to see if rmblastn existed somewhere I wasn’t looking:
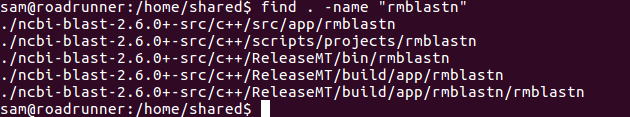
Additionally, I couldn’t find the location of the various BLAST executables. Some internet sleuthing led me to the NCBI page on installing BLAST+ from source, which indicates that the executables are stored in:
ncbi-blast-VERSION+-src/c++/ReleaseMT/bin/How intuitive! /s
In order to improve readability and usability of the /home/shared/ directory, I renamed the /home/shared/rmblast directory to reflect the BLAST version and created a symbolic link in that directory to the rmlbastn executable:
Symbolic link to RMBLAST

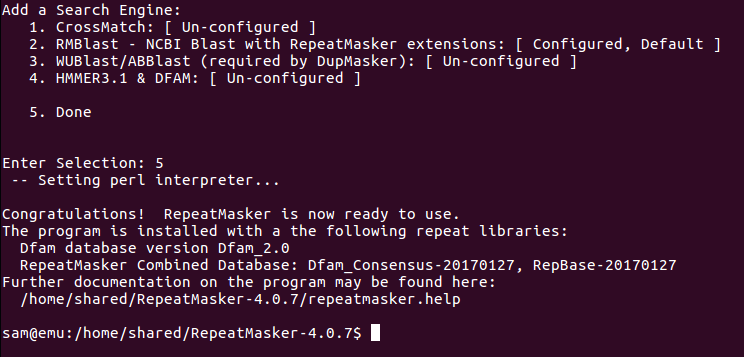
Initiate RepeatMasker configuration
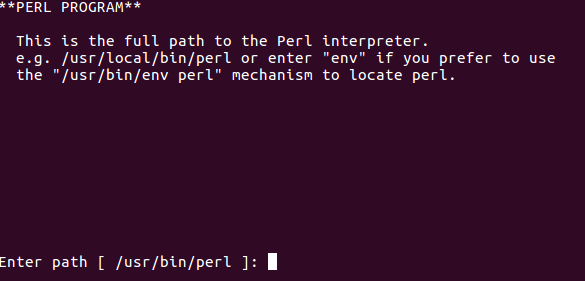
Confirm perl install location:
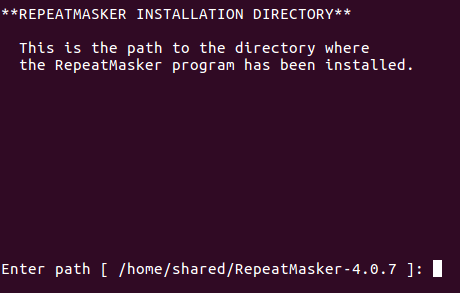
Confirm RepeatMasker install location:
Specify TRF install location:
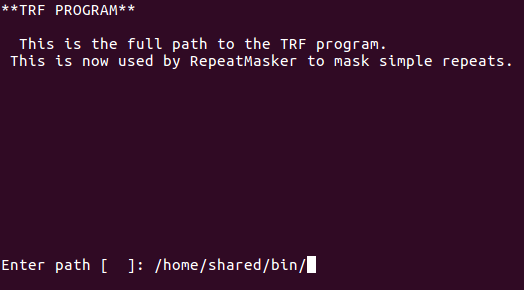
Hmmm, TRF error. Looking for file called trf:

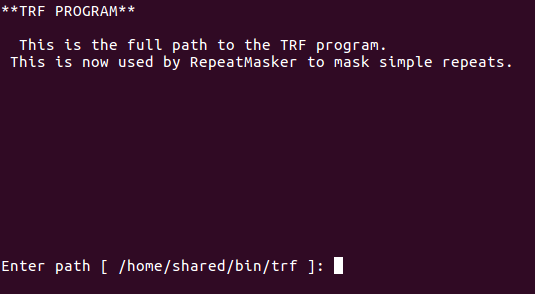
Renamed TRF file to trf and now it’s automatically found:
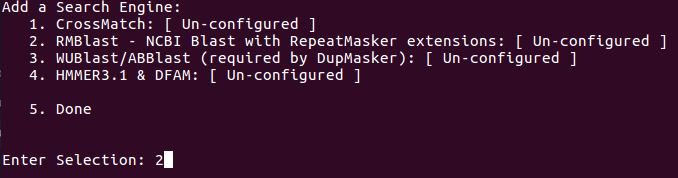
Set RMBlast as search engine:
Set RMBlast install location:
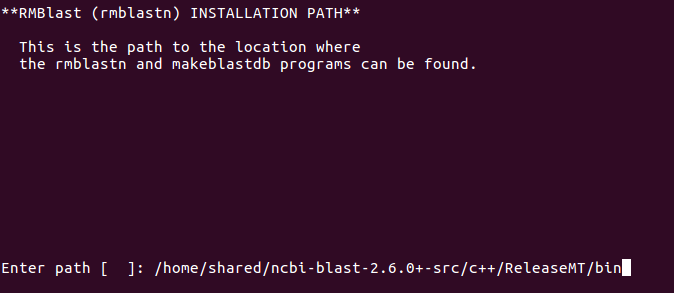
Set RMBlast as default search engine:
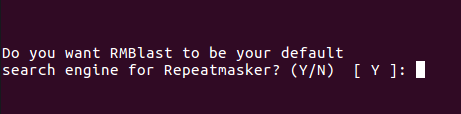
Confirmation of RMBlast as default search engine and successful installation of RepeatMasker: