I decided that I wanted to use the Olurida_v080 version instead (or, in addtion to?), as the Olurida_v080 version has not been size restricted (the Olurida v081 version is only contigs >1000bp). I feel like we could miss some important regions, so wanted to run this analysis using all of the genome data we currently have available. Additionally, this will be consistent with my previous Bismark (DNA methylation analysis).
Used HISAT2 on our HPC Mox node to align our RNAseq reads to our Olurida_v080 genome assembly:
SBATCH script file:
NOTE: For brevity sake, I have not listed all of the input RNAseq files below. Please see the full script, which is linked above.
#!/bin/bash
## Job Name
#SBATCH --job-name=20180926_oly_hisat2
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=5-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/20180926_oly_RNAseq_genome_hisat2_bedgraph
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
date >> system_path.log
echo "" >> system_path.log
echo "System PATH for $SLURM_JOB_ID" >> system_path.log
echo "" >> system_path.log
printf "%0.s-" {1..10} >> system_path.log
echo ${PATH} | tr : \\n >> system_path.log
# Set genome assembly path
oly_genome_path=/gscratch/srlab/sam/data/O_lurida/oly_genome_assemblies
# Set sorted transcriptome assembly bam file
oly_transcriptome_bam=20180926_Olurida_v080.sorted.bam
# Set hisat2 basename
hisat2_basename=Olurida_v080
# Set program paths
## hisat2
hisat2=/gscratch/srlab/programs/hisat2-2.1.0
## bedtools
bedtools=/gscratch/srlab/programs/bedtools-2.27.1/bin
## samtools
stools=/gscratch/srlab/programs/samtools-1.9/samtools
# Build hisat2 genome index
${hisat2}/hisat2-build \
-f ${oly_genome_path}/Olurida_v080.fa \
Olurida_v080 \
-p 28
# Align reads to oly genome assembly
${hisat2}/hisat2 \
--threads 28 \
-x "${hisat2_basename}" \
-q \
-1 \
-2 \
-S 20180926_"${hisat2_basename}".sam
# Convert SAM file to BAM
"${stools}" view \
--threads 28 \
-b 20180926_"${hisat2_basename}".sam > 20180926_"${hisat2_basename}".bam
# Sort BAM
"${stools}" sort \
--threads 28 \
20180926_"${hisat2_basename}".bam \
-o 20180926_"${hisat2_basename}".sorted.bam
# Index for use in IGV
##-@ specifies thread count; --thread option not available in samtools index
"${stools}" index \
-@ 28 \
20180926_"${hisat2_basename}".sorted.bam
# Create bedgraph
## Reports depth at each position (-bg in bedgraph format) and report regions with zero coverage (-a).
## Screens for portions of reads coming from exons (-split).
## Add genome browser track line to header of bedgraph file.
${bedtools}/genomeCoverageBed \
-ibam ${oly_transcriptome_bam} \
-bga \
-split \
-trackline \
> 20180926_oly_RNAseq.bedgraph
The script performs the following functions:
- Genome indexing
- RNAseq alignment to genome
- Convert SAM to BAM
- Sort and index BAM
- Determine RNAseq coverage
RESULTS
Output folder:
Bedgraph file (1.9GB):
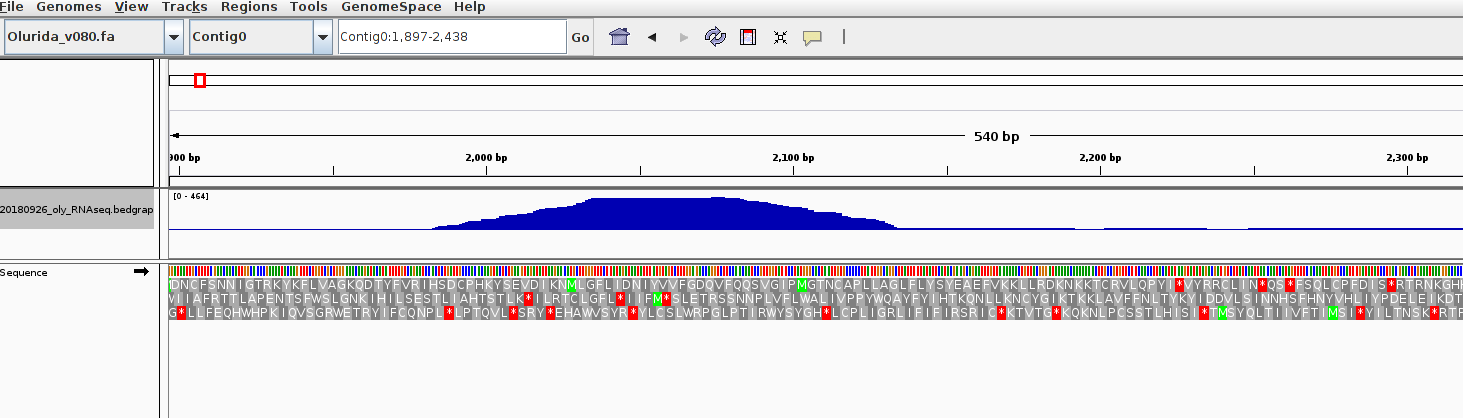
Loaded in to IGV to verify things looked OK: