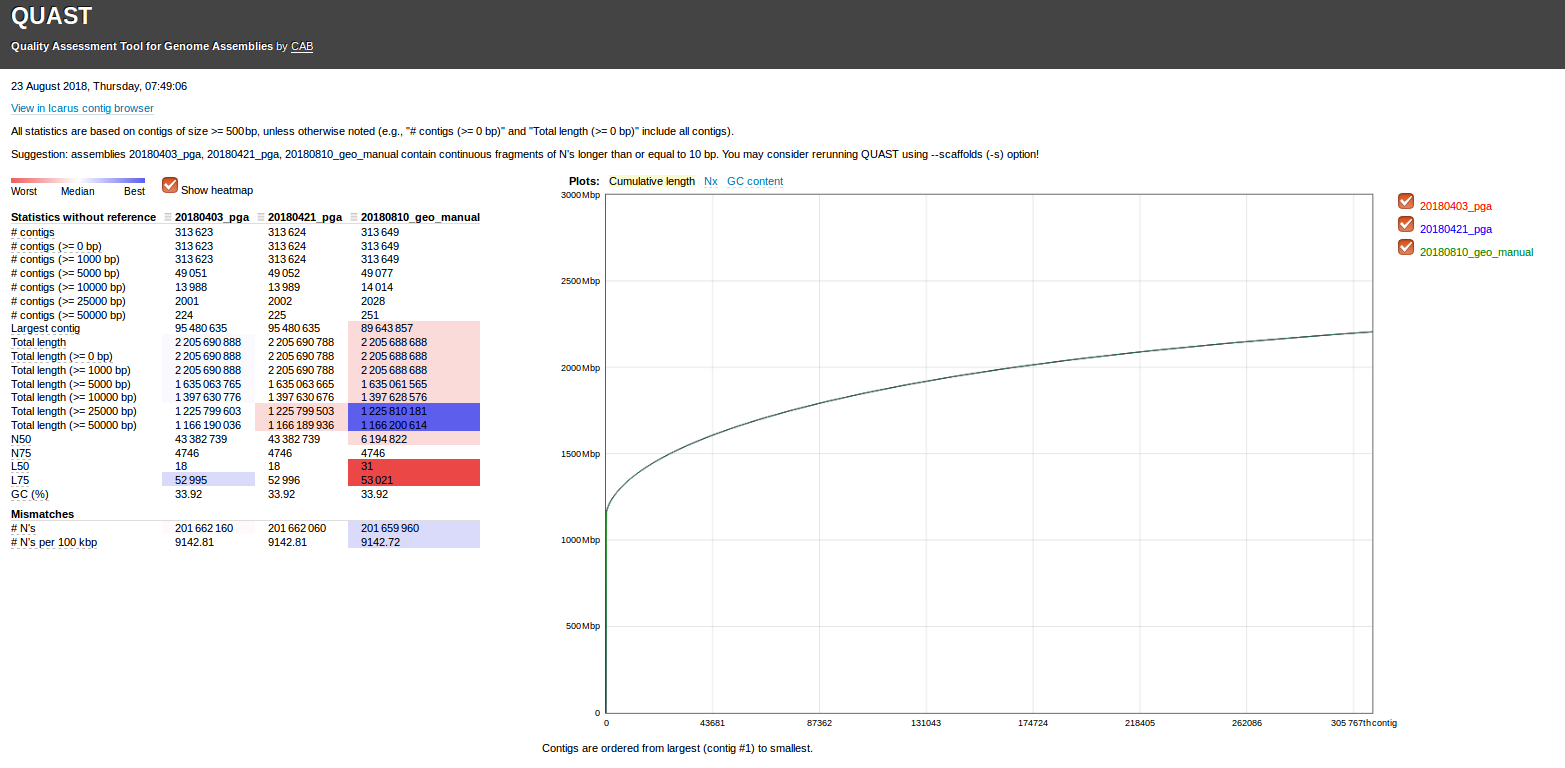
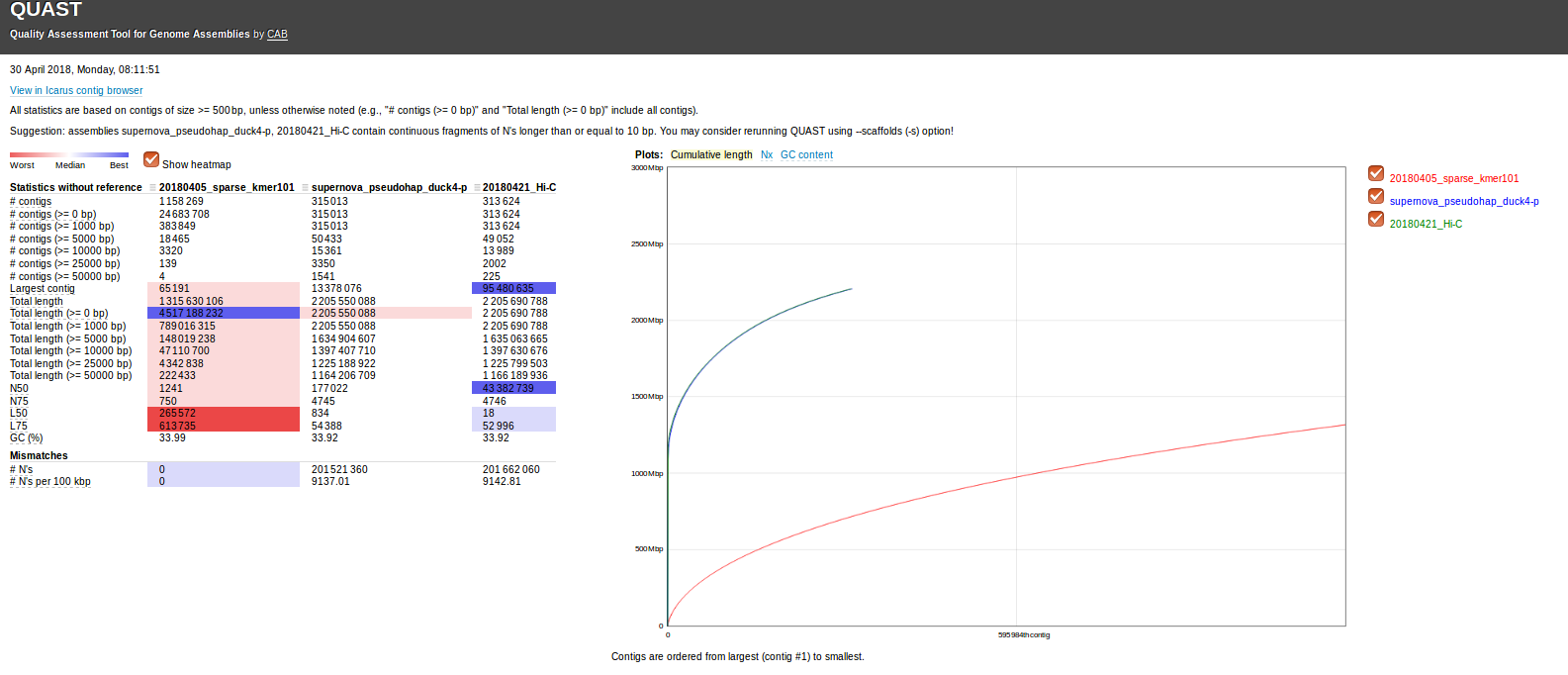
Steven came across a 2012 paper in BMC Bioinformatics (“Exploiting sparseness in de novo genome assembly”) that utilized an assembly program we hadn’t previously encountered: SparseAssembler
This software is intended to greatly reduce the required amount of RAM necessary to process very large assembly data sets. As I previously learned, RAM is a limiting factor for assembly programs, and the install (if you can even call it that) was simply upacking a zip file (program installations on Mox are not trivialso this seems like it has promise!
The job was run on our Mox node.
Here’s the batch script to initiate the job:
20180308_soap_novaseq_geoduck_slurm.sh
#!/bin/bash
## Job Name
#SBATCH --job-name=20180308_sparse_assembler_geo_novaseq
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes (We only get 1, so this is fixed)
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=30-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/20180308_SparseAssembler_novaseq_geoduck
/gscratch/srlab/programs/SparseAssembler/SparseAssembler
LD 0
NodeCovTh 1
EdgeCovTh 0
k 117
g 15
PathCovTh 100
GS 2200000000
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/AD002_S9_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/AD002_S9_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/AD002_S9_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/AD002_S9_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR005_S4_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR005_S4_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR005_S4_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR005_S4_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR006_S3_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR006_S3_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR006_S3_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR006_S3_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR012_S1_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR012_S1_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR012_S1_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR012_S1_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR013_AD013_S2_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR013_AD013_S2_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR013_AD013_S2_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR013_AD013_S2_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR014_AD014_S5_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR014_AD014_S5_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR014_AD014_S5_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR014_AD014_S5_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR015_AD015_S6_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR015_AD015_S6_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR015_AD015_S6_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR015_AD015_S6_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR019_S7_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR019_S7_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR019_S7_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR019_S7_L002_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR021_S8_L001_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR021_S8_L001_R2_001_val_2_val_2.fastq
i1 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR021_S8_L002_R1_001_val_1_val_1.fastq
i2 /gscratch/scrubbed/samwhite/20180129_trimmed_again/NR021_S8_L002_R2_001_val_2_val_2.fastq
Results
Output folder: 20180308_SparseAssembler_novaseq_geoduck/
Well, this failed, but not because of memory issues (which is a good start)!
Instead, it failed because the kmer size was too large??!!
See the slurm output log file:
Kmergenie had indicated a kmer size of 117bp.
Will reduce kmer size and try again. Fingers crossed…