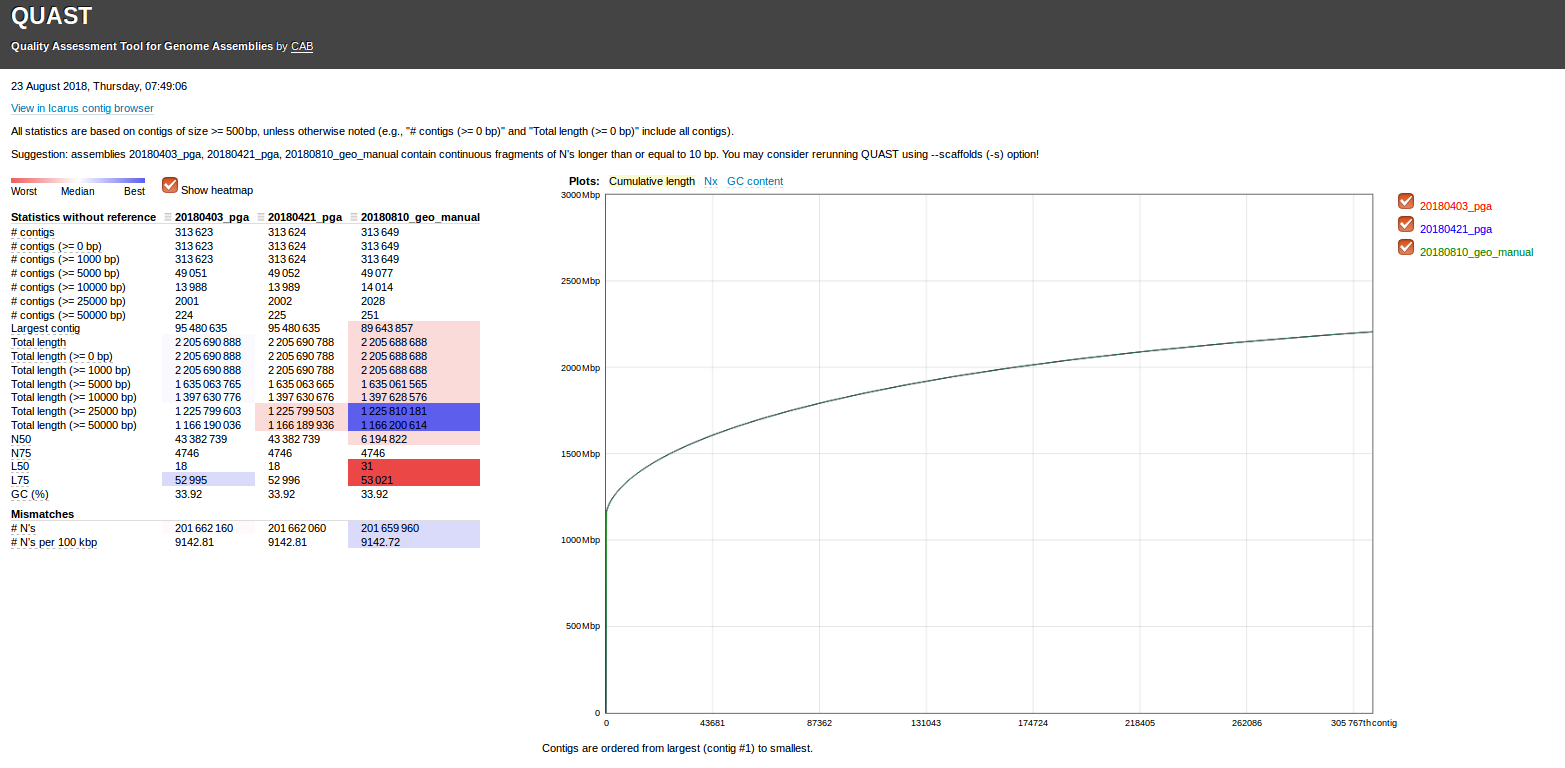
We received the final geoduck genome assembly data from Phase Genomics, in which they updated the assembly by performing some manual curation:
There are additional assembly files that provide some additional assembly data. See the following directory:
Actual sequencing data and two previous assemblies were previously received on 20180421.
All assembly data (both old and new) from Phase Genomics was downloaded in full from the Google Drive link provided by them and stored here on Owl:
Ran Quast to compare all three assemblies provided (command run on Swoose):
/home/sam/software/quast-4.5/quast.py \
-t 24 \
--labels 20180403_pga,20180421_pga,20180810_geo_manual \
/mnt/owl/Athaliana/20180421_geoduck_hi-c/Results/geoduck_roberts results 2018-04-03 11:05:41.596285/PGA_assembly.fasta \ /mnt/owl/Athaliana/20180421_geoduck_hi-c/Results/geoduck_roberts results 2018-04-21 18:09:04.514704/PGA_assembly.fasta \ /mnt/owl/Athaliana/20180822_phase_genomics_geoduck_Results/geoduck_manual/geoduck_manual_scaffolds.fasta
Results:
Quast output folder: results_2018_08_23_07_38_28/
Quast report (HTML): results_2018_08_23_07_38_28/report.html