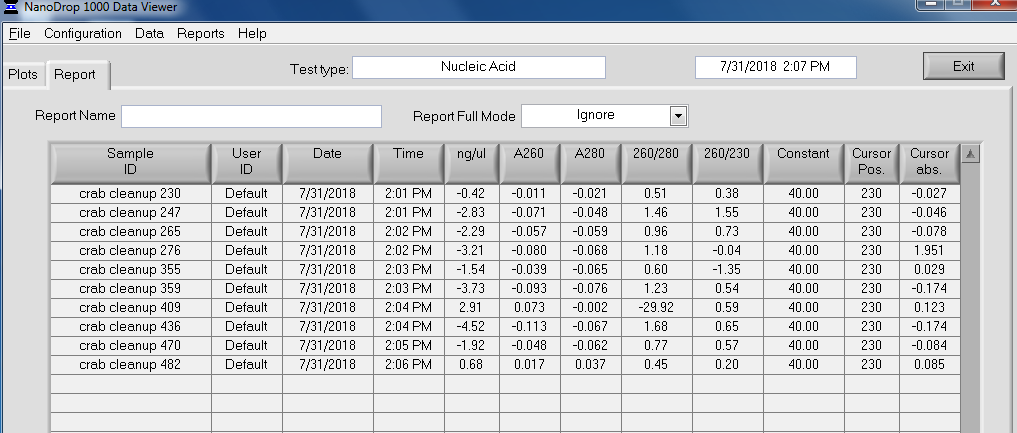
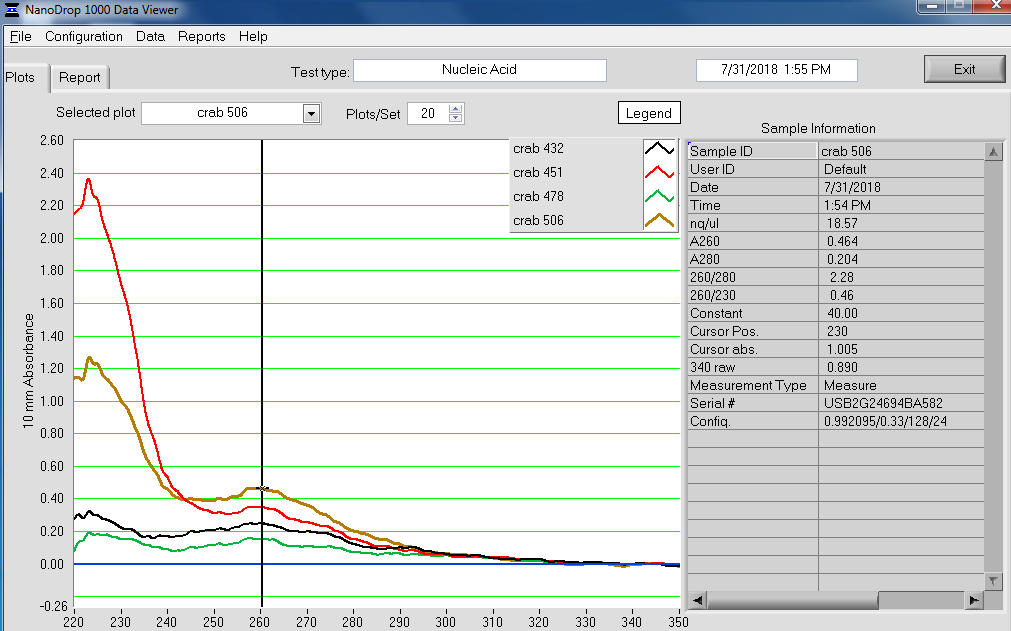
Made Illumina libraries with goeduck metagenome water filter DNA I previously isolated on:
We used a free Nextera DNA Flex Kit (Illumina) that we won in a contest held by Illumina!
Followed the manufacturer’s protocol for input DNA quantities <10ng with the following changes/notes:
- PCR steps performed in 200uL thin-walled PCR tubes.
-
Magnetic separations were performed in 1.7mL snap cap tubes.
-
Thermalcycler: PTC-200 (MJ Research)
-
Magnet: DynaMag 2 (Invitrogen)
See the Library Calcs sheet (link below) for original sample names and subsequent library sample names.
IMPORTANT!
The sheet also contains the indexes used for each library. This info will be necessary for sequencing facility.
Library Calcs (Google Sheet):
Links to the Illumina manuals are below:
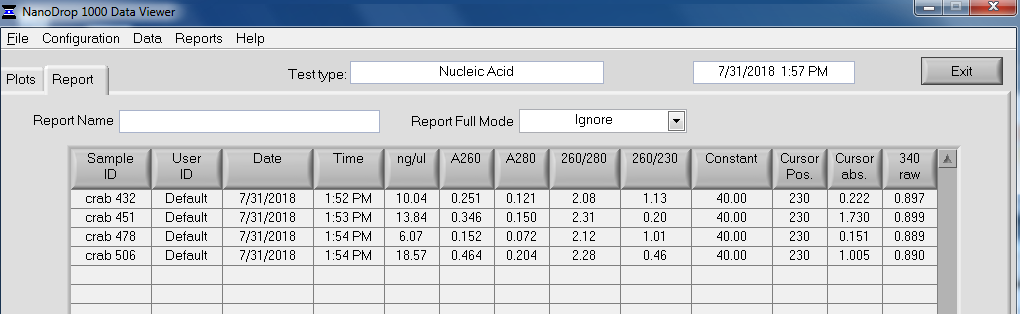
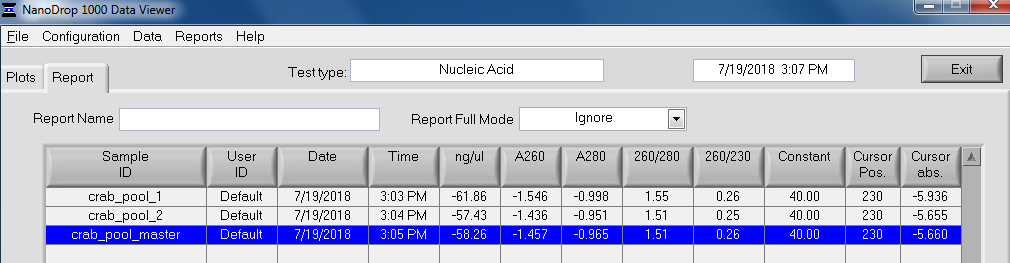
After library construction was completed, individual libraries were quantified on the Roberts Lab Qubit 3.0 (Invitrogen) with the Qubit 1x dsDNA HS Assay Kit.
2uL of each sample was used for each assay.
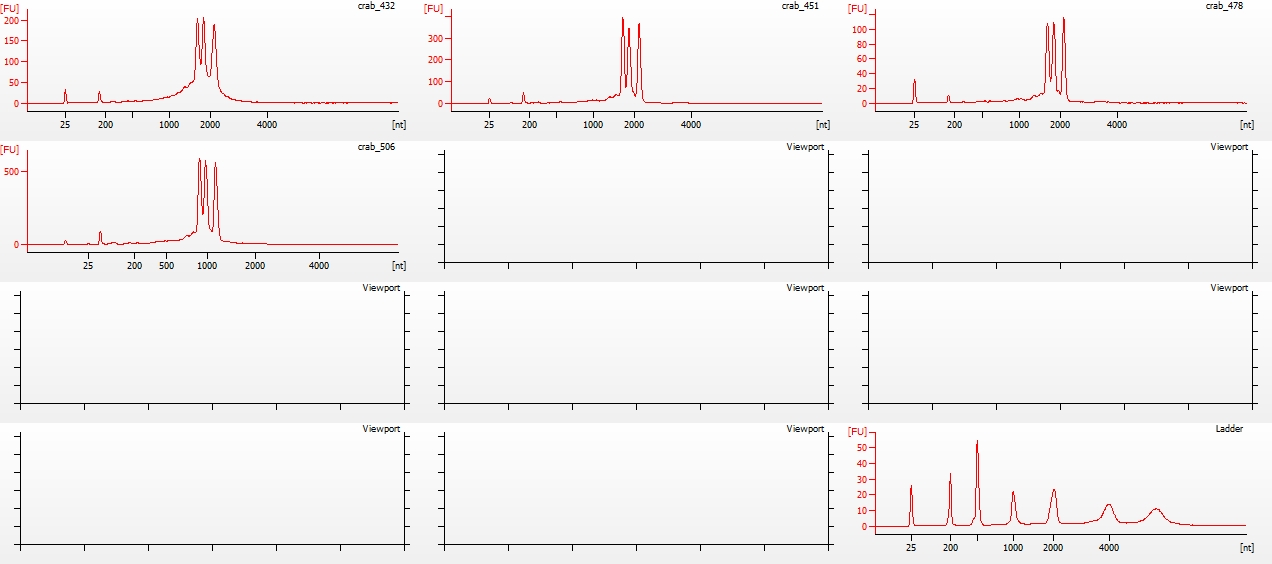
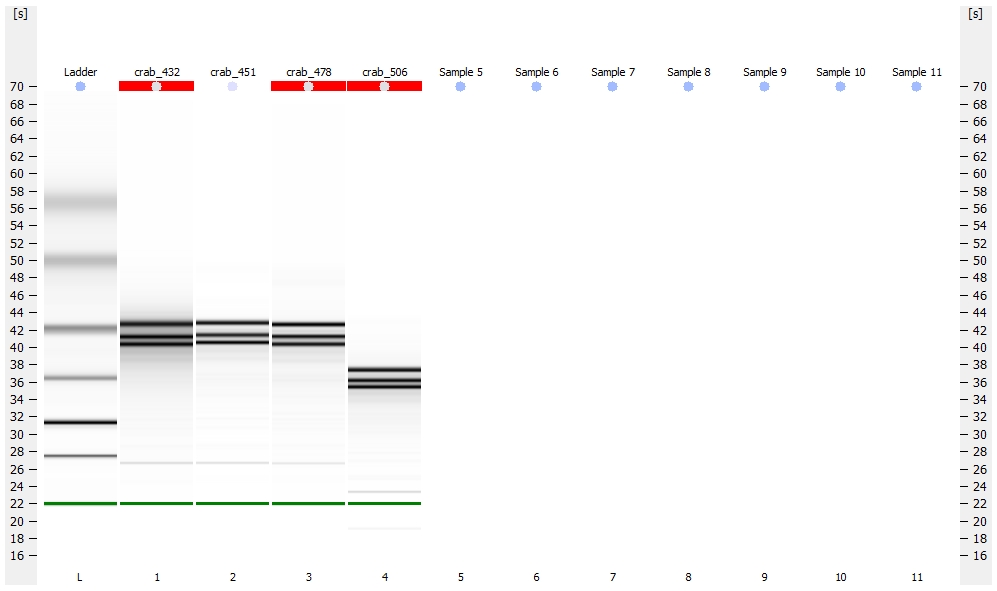
Library quality was assessed using the Seeb Lab 2100 Bioanalyzer (Agilent) with a High Sensitivity DNA Kit, using 1uL of each sample.
Libraries were stored in the small -20C in FTR213:
Results:
Qubit Raw Data (Google Sheet):
Bioanalyzer File (XAD):
All libraries have DNA in them, so that’s good!
Except for one library (Library Geoduck MG #04 is bad), the other libraries look OK (i.e. not great). Compared to the example on Pg. 12 in the manual, these libraries all have some extra high molecular weight stuff.
When selecting the range listed in the Nextera Kit manual, the average fragment size is ~530bp – the expected size should be ~600bp.
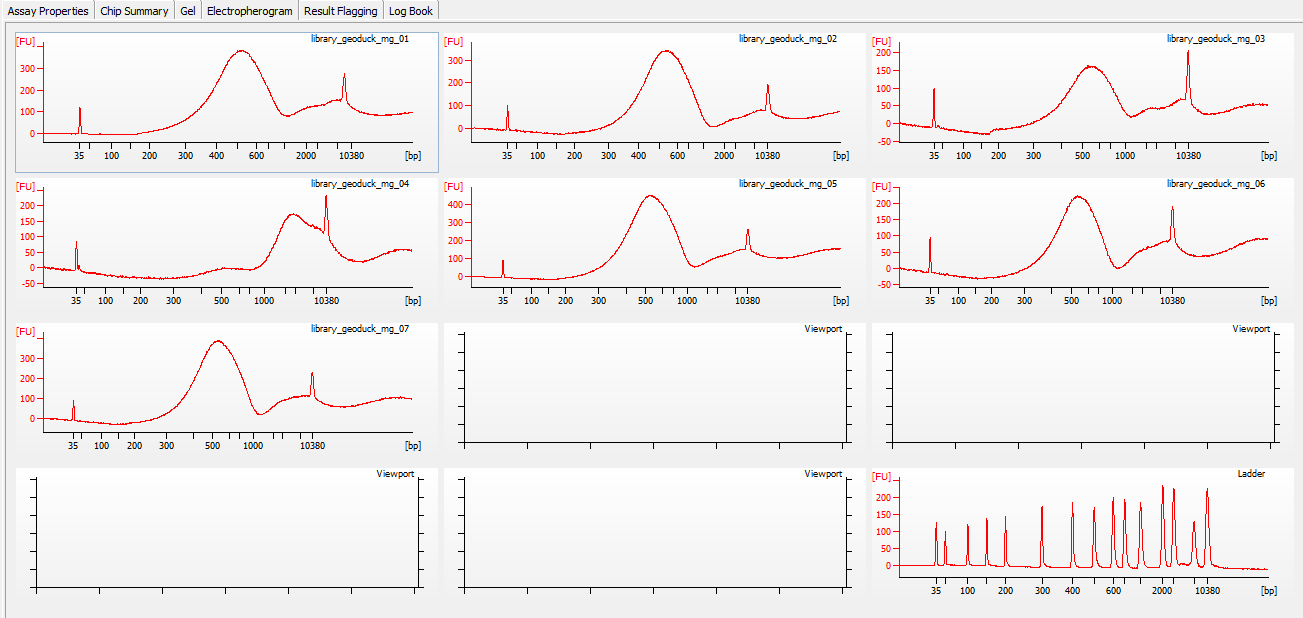
Spoke with Steven about Library Geoduck MG #04 and we’ve opted to just leave it out.
All other samples were pooled into a single samples according to the manufacturer’s protocol.
This pooled sample was stored in the same -20C box as above, in position I4.
UPDATE 20180808
After some confusion with the sequencing facility, I contacted Illumina regarding adapter sequences. I used the sequences provided for the Nextera DNA 24 CD Indexes (which was the index kit we used) on p.18 of the Illumina Index Adapter Pooling Guide.
As it turns out, these sequences are incorrect. The correct sequences are on p.12 of that document (the Nextera DNA 96 CD Indexes).
I’ve updated the Google Sheet (linked above) to reflect the correct index sequences.
Email from Illumina is below. Even though he specifically references the H705 adapter, the correct sequence information for all i7 index adapters is found on p.12.
Hi Sam,
Thanks for the clarification! For the index sequence H705, this sequence is incorrect in the Index Adapters Pooling Guide. The correct information is found on page 12 of the same document and should be:
H705 “AGGAGTCC” (Bases in Adapter) and “GGACTCCT” (bases for sample sheet.
This is also consistent with the Illumina Adapters letter.
We have provided this feed back to our colleagues to update the document so that all the information is consistent.
Thanks for your patience and understanding while we evaluated this issue. If we do have any other questions or concerns, please let us know and we would be happy to discuss this further.
Best,
Russell
Russell Chan, Ph.D.
Technical Applications Scientist
Illumina Technical Support
Telephone available 24 hours
Monday through Friday
Technical Bulletins: https://support.illumina.com/bulletins.html
Trainings: http://support.illumina.com/traidexes