Due to difficulties getting RNA from hemolymph samples stored in RNAlater, Grace is testing out lyophilizing samples before extraction. Who knows what impact this will have on RNA, but it’s worth a shot!
Isolated RNA from three crab hemolymph samples preserved in RNAlater (Test 1, Test 2, Test 3) that had been lyophilized overnight last week.
Samples were provided by Grace.
I believe the primary purpose for this particular test was to verify that the freeze dryer was a feasible tool, since Grace experienced a minor mishap when she attempted the lyohpilization initially.
Lyophilization was successful, without any mess.
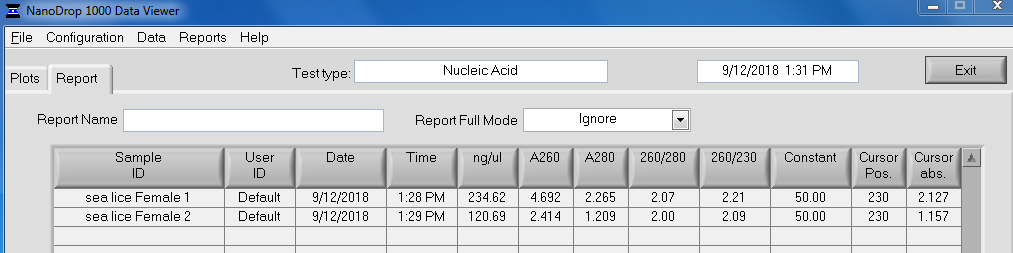
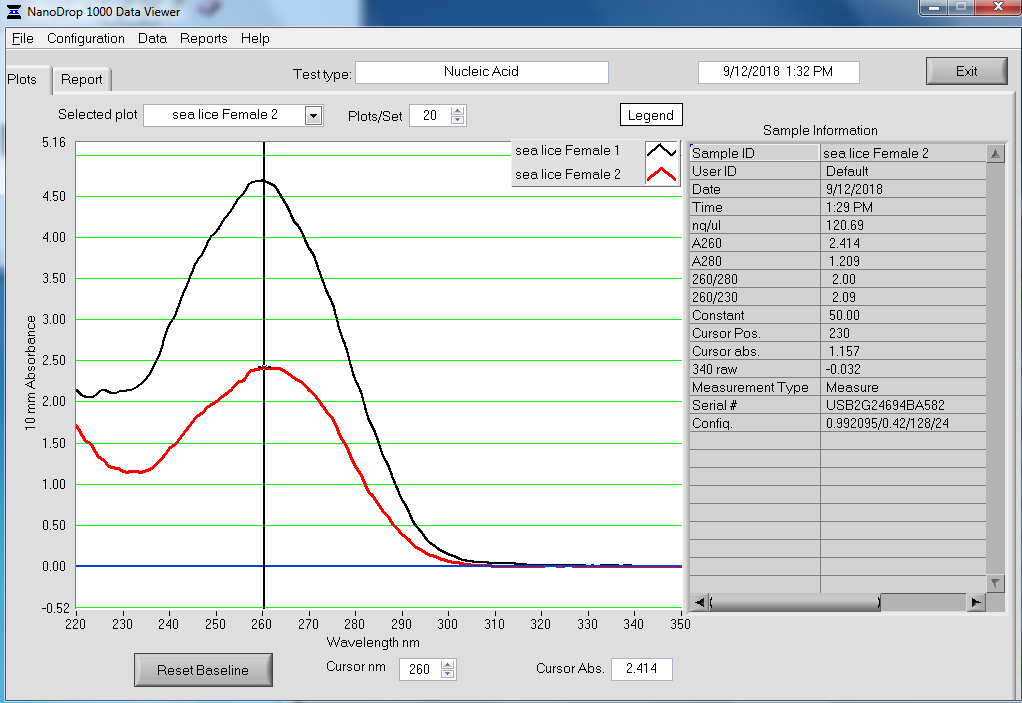
TEST 3 LYOPHILIZATION
Isolated RNA using TriReagent, according to manufacturer’s protocol:
Added 1mL TriReagent to each tube, vortexed to mix/dissolve solute, incubated 5mins at RT, added 200uL of chloroform, vortexed 15s to mix, incubated at RT for 5mins, centrifuged 15mins, 12,000g, 4oC, transferred aqueous phase to new tube, added 500uL isopropanol to aqueous phase, mixed, incubated at RT for 10mins, centrifuged 8mins, 12,000g, at RT, discarded supernatant, added 1mL 75% ethanol, centrifuged 5mins, 12,000g at RT, discarded supernatant and resuspended in 10uL of 0.1% DEPC-treated H2O.
Quantified RNA using Roberts Lab Qubit 3.0 with the Qubit RNA high sensitivity kit. Used 5uL of each sample.
RESULTS
Qubit (Google Sheet):
Only one sample (Test 3) had detectable levels of RNA (20.4ng/uL).
So, this little test demonstrates that RNA can be isolated from lyophilized samples and extracted with TriReagent. However, I have not evaluated RNA integrity on the Bioanalyzer. I think Grace has some additional samples she wanted to test this method on, so I think we’ll wait until there are more samples before we use the Bioanalyzer.
Will give sample to Grace for -80oC storage.