I’ll begin this entry with a TL;DR (becuase it’s definitely a very long read):
With that out of the way, here’s a fairly lengthy overview of what has been done to date with the titrator. In essence, this is a cumulative notebook entry of all the entries I should have been doing on a daily/weekly basis (I actually feel much shame for neglecting this – it’s a terrible practice and an even worse example for other lab members).
Teaser: there are graphs!
Anyway, I’ve spent a lot of time getting our titrator, protocols, and scripts to a point where we can not only begin collecting real data, but also actually analyze the data in a semi-automated way.
More recently, I’ve finally started taking some measurements to assess the consistency of the actual titrator and stumbled across an interesting observation that may (may not) have an impact on how we proceed with sample/data handling.
Protocols
Titrator SOP is the primary protocol that encompasses setting up/shutting down the titrator, use of the LabX software needed for recording/exporting data from the titrator, and how to implement the necessary scripts to handle the exported data is still in early stages.
In theory, the SOP should be rather straightforward, but due to the sensitivity involved with these measurements, the SOP needs to carefully address how to set things up properly, provide a means for documenting startup/shutdown procedures, and provide troubleshooting assistance (e.g. how to empty/remove burette if/when air bubbles develop).
Overall, it’s a bit of a beast, albeit and important one, but I’ve put it on the back burner in order to focus my efforts/time on getting to the point of being able to collect data from the titrator and feel confident that we’re getting good readings.
Once I get to that point and am able to begin running samples, I’ll be able to dedicate more time to fleshing out the SOP, including adding pictures of all the components.
Scripts
parsing_TA_output.R is the script that has consumed the majority of my titrator-related time in the last couple of weeks. It is fully functional (it only requires manual entry of the exported LabX data file location). However, I’m hoping to eventually automate this as well – i.e. when new LabX export file appears, this script will execute. I won’t be spending much time on this aspect of the script until I
This has been my highest priority. Without having this script in a usable state, it has been a MAJOR slog to manually retrieve the appropriate data necessary to use in TA determination.
This also has been my biggest challenge with the titrator process. Here are just some of the hurdles I’ve had to deal with in putting this script together:
- “learning” R
- handling “dynamic” titrator output data
- this is not an easy task for a non-programmer!
- the output data is of differing numbers of rows from sample to sample, so the script had to be able to handle this aspect automatically
- making the script “flexible” (i.e. no “magic numbers”) to handle any number of samples without the user having to manually modify the script
- making th script “flexible” by operating on column names instead of column numbers, since column numbers were/are not constant, depending on changes to the script
- calculations resulting from two-part titration
- still not sure if I ever would’ve figured this out if I hadn’t taken the intro computer science class at UW a couple of years ago!
Despite all of this, I also feel like it’s one of my biggest accomplishments! It’s super satisfying to have this script functioning with virtually no user input required!
pH_calibration_check.R is still a work in progess, but is easily usable. Currently, it still has some hard-coded values (row numbers) in it for parsing data, but that should be easy to fix after what I went through with the TA parsing script!
Eventually, these two scripts will work in tandem, with the pH_calibration_check script exporting data to a daily “log” file, which the parsing_TA_output script will use to read-in the necessary pH data.
TA_calculation.R will calculate the TA values, but currently requires fully manual data entry. It desperately needs attention and will likely be my primary focus in the immediate future, due to the need to have TA values for actual samples, as well as daily quality control checks (e.g. verify CRM measurements look OK before measuring actual samples).
Measurements
Consistency checks with Instant Ocean
Instant Ocean Tests
I ran nine replicates of Instant Ocean (36g/L in deionized water) at two different samples weights/volume (50g, 75g) to make sure the titrator was producing consistent results.
Here’s the R Studio Project folder with all the data/scripts used to gather the data and produce the plots:
TA values were determined using the seacarb R package. I used a salinity of 35 (seacarb default value?), but this has not been determined for this batch of Instant Ocean.

| Sample Volume |
Mean TA |
Standard Deviation |
| 50mL |
669.4 |
11.14 |
| 75mL |
645.0 |
11.96 |
The first thing I noticed was the low TA values when using Instant Ocean. I expected these to be more similar to sea water, but the Instant Ocean hasn’t been aerated, so maybe that could account for the low TA values. Regardless, this shouldn’t be too much of an issue, since I only wanted to use this to see if we were getting consistent measurements.
The second thing I noticed was the difference in TA values between the 50mL and 75mL samples. This is/was odd, as sample weight is taken into account with the seacarb package.
So, I decided to explore this a bit further, using the CRMs that we have. I felt that this would provide more informative data regarding measurement accuracy (i.e. do our measurements match a known value?), in addition to further evaluation of the effects of sample volume on TA determination.
Consistency checks with CRMs
CRM Tests
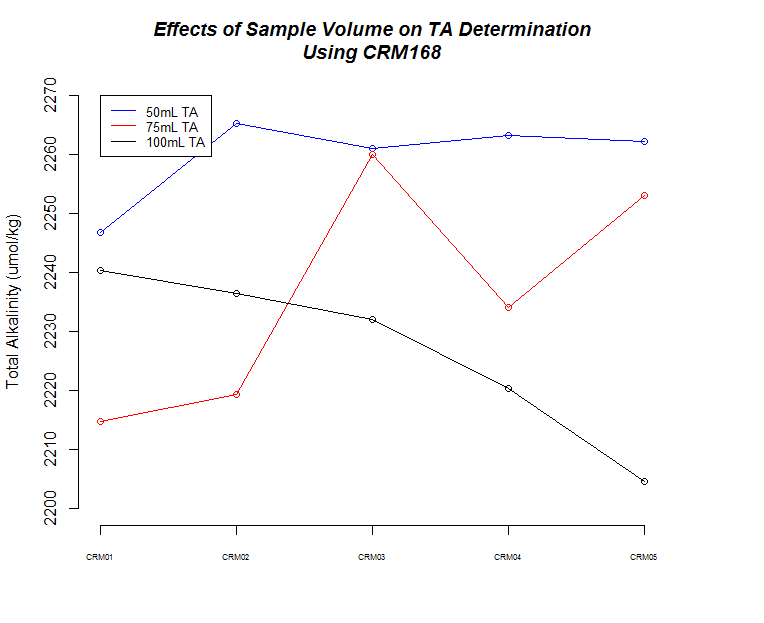
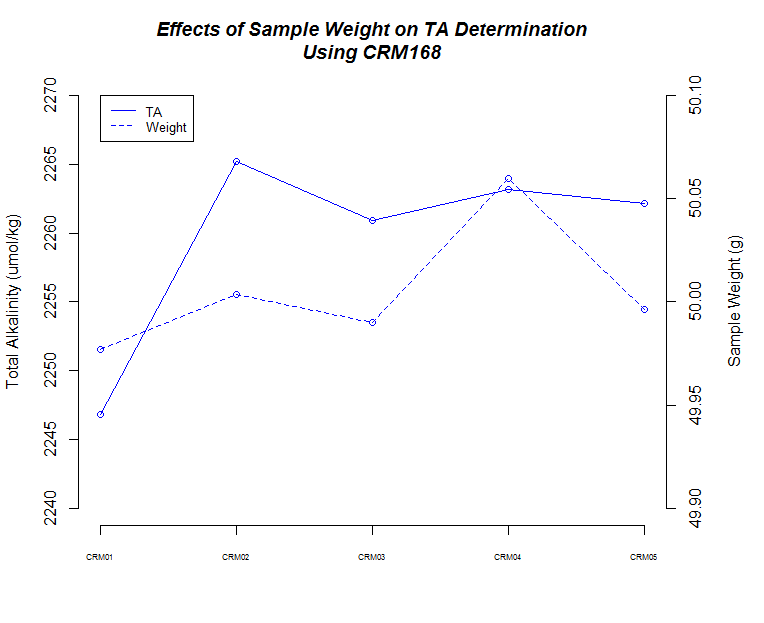
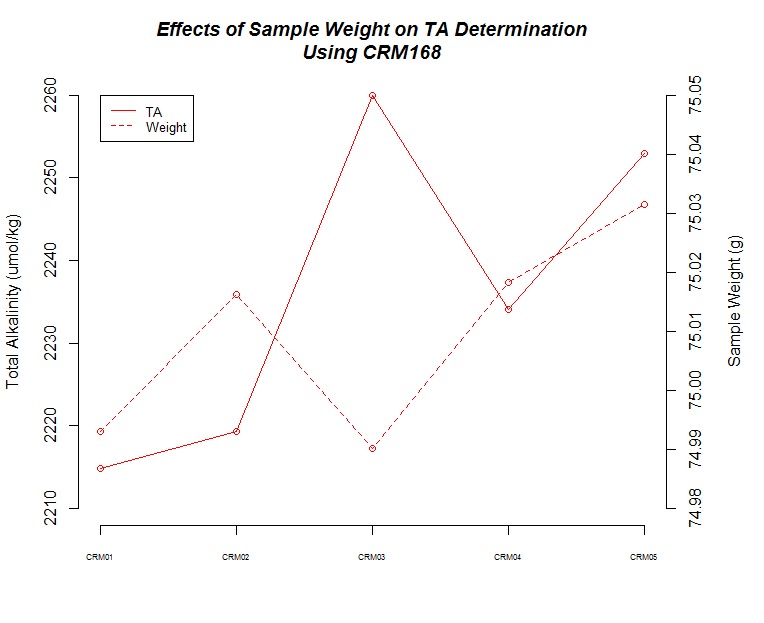
I ran five replicates of CRM Batch 168 (PDF) at three different sample weights/volume (50g, 75g, 100g) to make sure the titrator was producing consistent results and evaulate how accurate our measurements are.
Here’s the R Studio Project folder with all the data/scripts used to gather the data and produce the plots:
Here’s a bunch of graphs to consider:

| Sample |
Mean TA |
Standard Deviation |
| CRM 168 |
2071.47 |
NA |
| 50mL |
2259.66 |
7.35 |
| 75mL |
2236.22 |
19.96 |
| 100mL |
2226.73 |
14.49 |
First thing to notice is that all sample measurements, regardless of volume, produce a TA value that is ~10% higher than what the CRM is certified to be. I’ve previously discussed this with Hollie and she’s indicated that there are two options:
- Calculate an offset relative to what the CRM is supposed to be and apply this offset to any sample measurements.
-
Do not determine offset and just report calculated values, while providing CRM info.
The next thing that I noticed is the 50mL (g) samples produced the most consistent measurements.
There also seems to be a pattern where fluctuations in TA values across replicates are mirrored by changes in weight for each corresponding replicate.
Finally, although it isn’t explicitly addressed, there is a time element in play here. As sample number increases, the longer those samples sat in the sample changer before titration. Oddly, it appears that there could be an effect on samples as they sit (e.g. sample evaporation prior to titration) when one considers the 75mL and 100mL samples, but both of those result in opposing trends, while the 50mL samples do not seem to suffer from any sort of time-related changes…
The Wrap Up
Whew! We made it! I’ll wait to get some feedback from lab members and Hollie before cranking through all of Hollie’s samples, but I feel pretty good about proceeding with a 50mL sample volume. If we decide to calculate an offset later on, it should only be a relatively minor tweak to our script.
Next up, figure out a way to pull out all of Hollie’s salinity data for the samples I’m going to measure and incorporate that into the TA_calculation.R script.