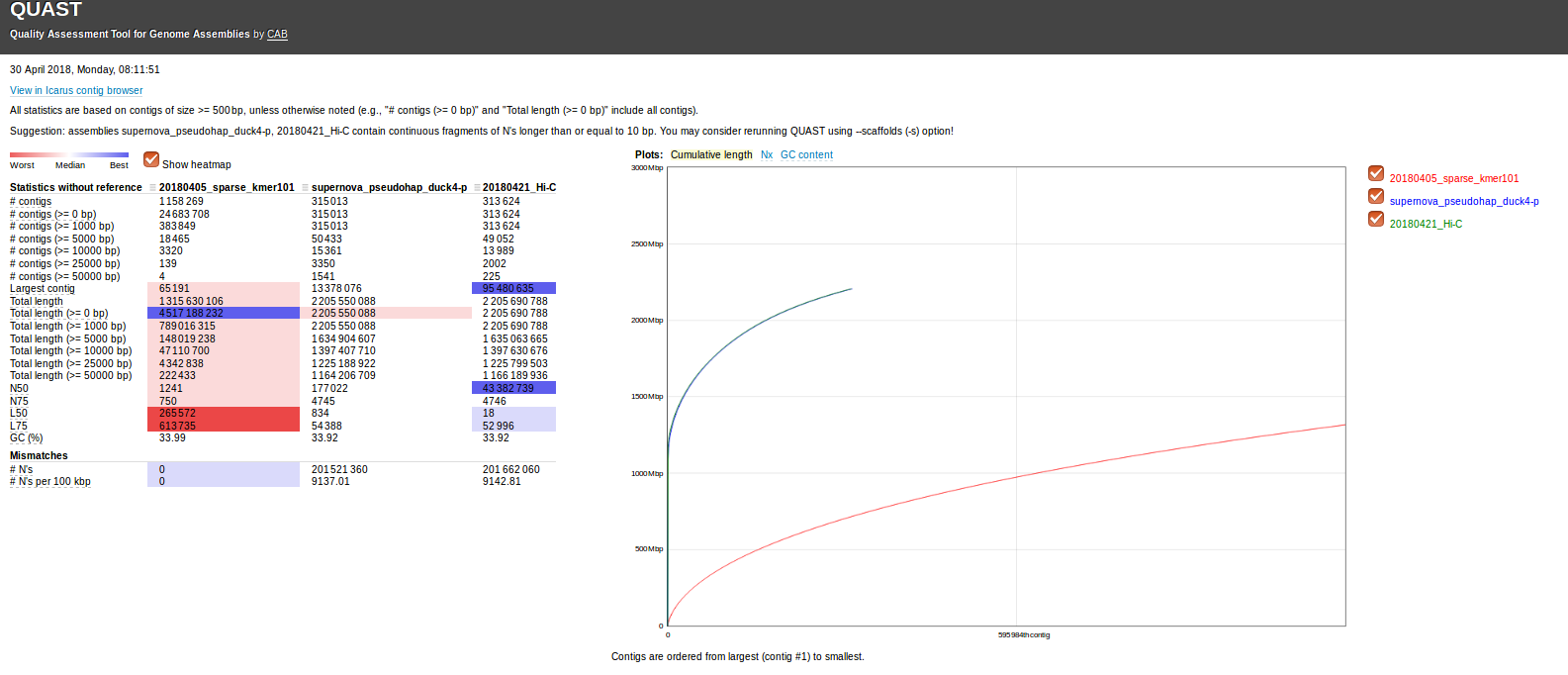
As part of the Illumina collaborative geoduck genome sequencing project, their end goal has always been to sequence the genome in a single run.
They’ve finally attempted this by running 10x Genomics, Hi-C, Nextera, and TruSeq libraries in a single run of the NovaSeq.
I downloaded the data using the BaseSpace downloader using Chrome on a Windows 7 computer (this is not available on Ubuntu and the command line tools that are available from Illumina are too confusing for me to bother spending the time on to figure out how to use them just to download the data).
Data was saved here:
Generated MD5 checksums (using md5sum on Ubuntu) and appended to the checksums file:
Illumina was unable to provide MD5 checksums on their end, so I was unable to confirm data integrity post-download.
Illumina sample info is here:
Will add info to:
List of files received:
10x-Genomics-Libraries-Geo10x5-A3-MultipleA_S10_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleA_S10_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleA_S10_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleA_S10_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleB_S11_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleB_S11_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleB_S11_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleB_S11_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleC_S12_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleC_S12_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleC_S12_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleC_S12_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleD_S13_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleD_S13_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleD_S13_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x5-A3-MultipleD_S13_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleA_S14_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleA_S14_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleA_S14_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleA_S14_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleB_S15_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleB_S15_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleB_S15_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleB_S15_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleC_S16_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleC_S16_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleC_S16_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleC_S16_L002_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleD_S17_L001_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleD_S17_L001_R2_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleD_S17_L002_R1_001.fastq.gz 10x-Genomics-Libraries-Geo10x6-B3-MultipleD_S17_L002_R2_001.fastq.gz HiC-Libraries-GeoHiC-C3-N701_S18_L001_R1_001.fastq.gz HiC-Libraries-GeoHiC-C3-N701_S18_L001_R2_001.fastq.gz HiC-Libraries-GeoHiC-C3-N701_S18_L002_R1_001.fastq.gz HiC-Libraries-GeoHiC-C3-N701_S18_L002_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP10-B2-AD013_S7_L001_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP10-B2-AD013_S7_L001_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP10-B2-AD013_S7_L002_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP10-B2-AD013_S7_L002_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP11-C2-AD014_S8_L001_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP11-C2-AD014_S8_L001_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP11-C2-AD014_S8_L002_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP11-C2-AD014_S8_L002_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP12-D2-AD015_S9_L001_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP12-D2-AD015_S9_L001_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP12-D2-AD015_S9_L002_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP12-D2-AD015_S9_L002_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP9-A2-AD002_S6_L001_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP9-A2-AD002_S6_L001_R2_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP9-A2-AD002_S6_L002_R1_001.fastq.gz Nextera-Mate-Pair-Library-GeoNMP9-A2-AD002_S6_L002_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA1-A1-NR006_S1_L001_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA1-A1-NR006_S1_L001_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA1-A1-NR006_S1_L002_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA1-A1-NR006_S1_L002_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA3-C1-NR012_S2_L001_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA3-C1-NR012_S2_L001_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA3-C1-NR012_S2_L002_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA3-C1-NR012_S2_L002_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA5-E1-NR005_S3_L001_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA5-E1-NR005_S3_L001_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA5-E1-NR005_S3_L002_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA5-E1-NR005_S3_L002_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA7-G1-NR019_S4_L001_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA7-G1-NR019_S4_L001_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA7-G1-NR019_S4_L002_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA7-G1-NR019_S4_L002_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA8-H1-NR021_S5_L001_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA8-H1-NR021_S5_L001_R2_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA8-H1-NR021_S5_L002_R1_001.fastq.gz Trueseq-stranded-mRNA-libraries-GeoRNA8-H1-NR021_S5_L002_R2_001.fastq.gz