After the last SparseAssembler assembly completed, I wanted to do another run with a different kmer size (last time was arbitrarily set at 101). However, I didn’t really know how to decide, particularly since this assembly consisted of mixed read lenghts (50bp and 100bp). So, I ran kmergenie on all of our geoduck (Panopea generosa) sequencing data in hopes of getting a kmer determination to apply to my next assembly.
The job was run on our Mox HPC node.
Slurm script: 20180419_kmergenie_geoduck_slurm.sh
Input files list (needed for kmergenie command – see Slurm script linked above): geoduck_fastq_list.txt
Results:
Output folder: 20180419_kmergenie_geoduck/
Slurm output file: 20180419_kmergenie_geoduck/slurm-161551.out
Kmer histograms (HTML): 20180419_kmergenie_geoduck/histograms_report.html
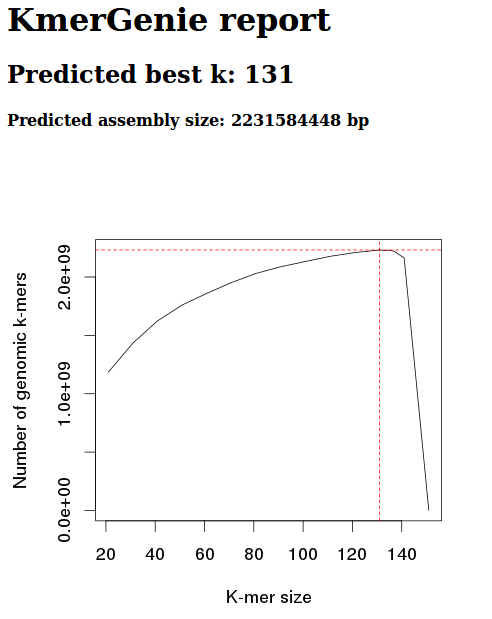
Screen cap from Kmer report:
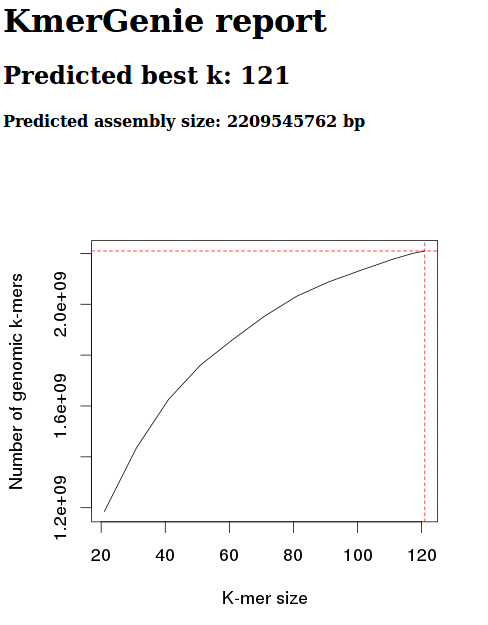
This data estimates the best kmer size for this data to be 121.
However, based on the kmergenie documentation, this is likely to be inaccurate. This inaccuracy is based on the fact that our kmer graph should be concave. Our graph, instead, is only partial – we haven’t reached a kmer size where the number of kmers is decreasing.
As such, I’ll try re-running with a different maximum kmer settting (default max is 121).