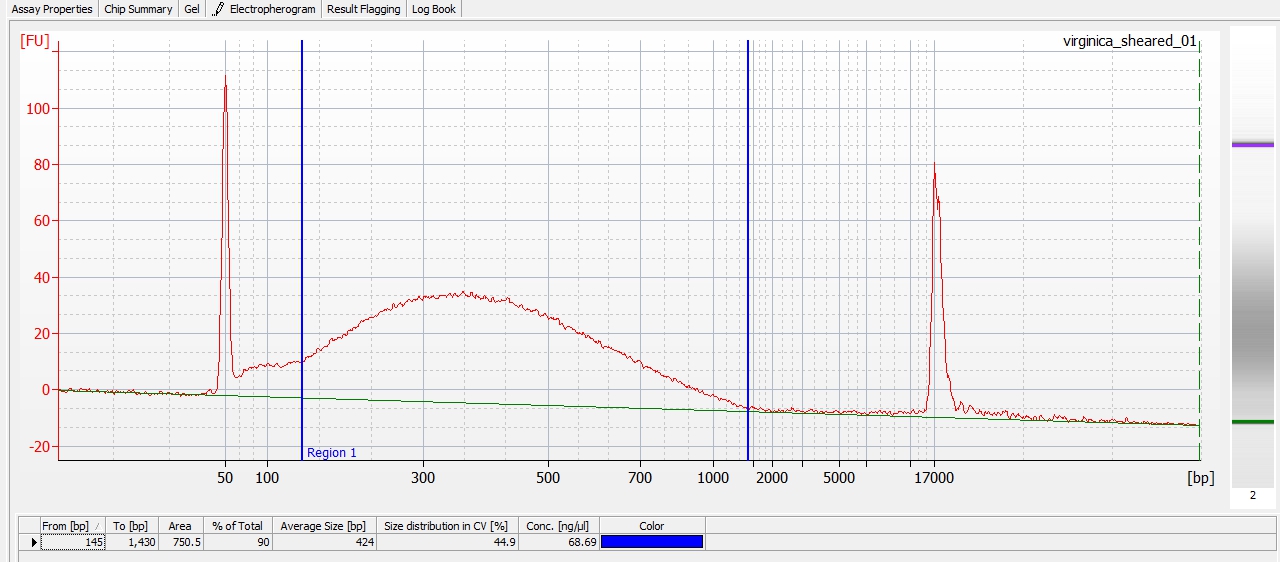
Here are the things I plan to tackle throughout the month of May:
Geoduck Reproductive Development Transcriptomics
My primary goal for this project is to successfully isolate RNA from the remaining, troublesome paraffin blocks that have yet to yield any usable RNA. The next approach to obtain usable quantities of RNA is to directly gouge tissue from the blocks instead of sectioning the blocks (as recommended in the PAXgene Tissue RNA Kit protocol). Hopefully this approach will eliminate excess paraffin, while increasing the amount of input tissue. Once I have RNA from the entire suite of samples, I’ll check the RNA integrity via Bioanalyzer and then we’ll decide on a facility to use for high-throughput sequencing.
BS-Seq Illumina Data Assembly/Mapping
Currently, there are two projects that we have performed BS-Seq with (Crassostrea gigas larvae OA (2011) bisulfite sequencing and LSU C.virginica Oil Spill MBD BS Sequencing) and we’re struggling to align sequences to the C.gigas genome. Granted, the LSU samples are C.virginica, but the C.gigas larvae libraries are not aligning to the C.gigas genome via standard BLASTn or using a dedicated bisulfite mapper (e.g. BS-Map). I’m currently BLASTing a de-novo assembly of the C.gigas larvae OA 400ppm sequencing that Steven made against the NCBI nt DB in an attempt to assess the taxonomic distribution of the sequences we received back. I’ll also try using a different bisulfite mapper, bismark, that Mackenzie Gavery has previously used and has had better results with than BS-Map.
C.gigas Heat Stress MeDIP/BS-Seq
As part of Claire’s project, there’s still some BS-Seq data that would be nice to have to complement the data she generated via microarray. It would be nice to make a decision about how to proceed with the samples. However, part of our decision on how to proceed is governed by the results we get from the two projects above. Why do those two projects impact the decision(s) regarding this project? They impact this project because in the two projects above, we produced our own BS-Seq libraries. This is extremely cost effective. However, if we can’t obtain usable data from doing the library preps in-house, then that means we have to use an external service provider. Using an external company to do this is significantly more expensive. Additionally, not all companies can perform bisulfite treatment, which limits our choices (and, in turn, pricing options) on where to go for sequencing.
Miscellany
When I have some down time, I’ll continue working on migrating my Wikispaces notebook to this notebook. I only have one year left to go and it’d be great is all my notebook entries were here so they’d all be tagged/categorized and, thus, be more searchable. I’d also like to work on adding README files to our plethora of electronic data folders. Having these in place will greatly facilitate the ability of people to quickly and more easily figure out what these folders contain, file formats within those folders, etc. I also have a few computing tips/tricks that I’d like to add to our Github “Code” page. Oh, although this isn’t really lab related, I was asked to teach the Unix shell lesson (or, at least, part of it) at the next Software Carpentry Workshop that Ben Marwick is setting up at UW in early June. So, I’m thinking that I’ll try to incorporate some of the data handling stuff I’ve been tackling in lab in to the lesson I end up teaching. Additionally, going through the Software Carpentry materials will help reinforce some of the “fundamental” tasks that I can do with the shell (like find, cut and grep).
In the lab, I plan on sealing up our nearly overflowing “Broken Glass” box and establishing a new one. I need to autoclave, and dispose of, a couple of very full biohazard bags. I’m also going to vow that I will get Jonathan to finally obtain a successful PCR from his sea pen RNA.